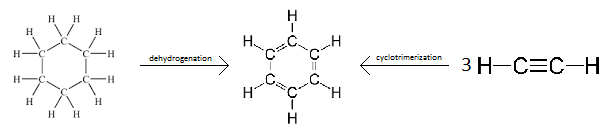
Aromatization
Aromatization is a chemical reaction in which an aromatic system is formed. It can also refer to the production of a new aromatic moiety in a molecule which is already aromatic. Theoretically, this can be achieved by dehydrogenation of existing cyclic compounds (such as in converting cyclohexane into benzene) or by formation of new cyclic system (such as in the cyclotrimerization of acetylene to benzene);[1] practically, other moieties are typically required to carry out such conversion, and other approaches like applying condensation reactions are possible. Aromatization includes the formation of any aromatic system (including heterocyclic systems), and is not restricted to benzene and its derivatives.
History
Benzene
The substance now called benzene, C6H6, was known as a component of Southeast Asian aromatic resins used in perfumery since the 15th century.[2] It was first isolated and identified by Michael Faraday in 1825 who named it bicarburet of hydrogen.[3][4] In 1845, Charles Mansfield, working under August Wilhelm von Hofmann, isolated benzene from coal tar and began the industrial-scale production based on this method four years later.[5][6] Over time, consensus developed that other substances were chemically related to benzene, comprising a diverse chemical family; Hofmann used the word "aromatic" for this family for the first time in 1856.[7]

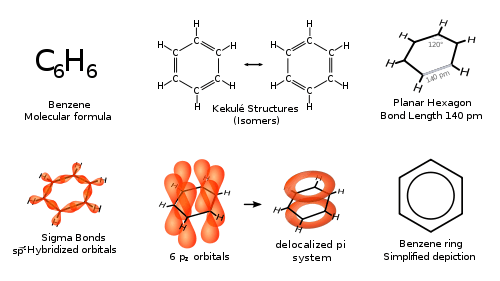
Historic interest in benzene arose from the uncertainty over its structure and reactivity. It was well established that it had a carbon to hydrogen ratio of 1:1 which suggested the presence of double or triple bond carbon-to-carbon bonds, yet such unsaturated compounds typically undergo addition reactions, which benzene does not. Numerous potential structures were proposed, including by Claus,[8] Dewar,[9] Ladenburg,[10] Armstrong,[11] and Thiele.[12] The most influential proposal was that of Kekulé who in 1865 suggested a cyclic structure of six carbon atoms with alternating single and double bonds[13] Kekulé based his argument on the now well-known observations of arene substitution patterns, namely that there are always only one isomer of any monoderivative of benzene and three isomers of every disubstituted derivative.[14] Critics noted that Kekulé's proposal implied that there should be two distinguishable isomers for an ortho-substitution (a 1,2-disubstituion) depending on whether a single or double bond joined these two carbons. Kekulé suggested in reply that benzene had two complementary structures and that these forms rapidly interconverted, which would also explain the lack of addition reactions (as the valency of each carbon atom is not that expected of a double bond). Though this interconversion is incorrect —in fact, they are both resonance contributors to the actual structure (which is closest to Thiele's structure)— the geometry and structure proposed by Kekulé are correct. Benzene is the prototypical example of an aromatic compound, a hitherto unknown class of compounds, and its synthesis was the first example of aromatization, the process and chemical reactions whereby an aromatic system is formed. The new understanding of benzene and all aromatic compounds was so important for both pure and applied chemistry that in 1890 the German Chemical Society organized an elaborate appreciation in Kekulé's honor, celebrating the twenty-fifth anniversary of his first benzene paper;[15] its 150th anniversary was also marked.[16] The cyclic nature of benzene was confirmed crystallographically by Kathleen Lonsdale in 1929.[17][18]
X-ray diffraction confirms that all six carbon-carbon bonds in benzene are the same length, 140 pm (1.40 Å) – intermediate between the bond lengths of single (154 pm) and double (134 pm) carbon-carbon bonds.[19] This is consistent with delocalization of the electrons from the double bonds in Kekulé's structures equally between each of the six carbon atoms which form a planar ring.[20] The molecular orbital theory description involves the formation of three delocalized π orbitals spanning all six carbon atoms, while the valence bond theory description involves a superposition of resonance structures.[21][22] This electronic structure provides benzene with unusual stability and contributes to the molecular and chemical properties which are now known as aromaticity. To accurately reflect the nature of the bonding as having neither single nor double bonds, but rather something in between, benzene is often depicted with a circle inside a hexagonal arrangement of carbon atoms, similar to the structure first illustrated by Thiele.[12]
Aromaticity

In organic chemistry, the term aromaticity has come to refer to cyclic (ring-shaped), planar (flat) organic compounds with a ring of resonance bonds that exhibit unusually high stability relative to other geometric or connective arrangements with the same set of atoms.[24] Aromatic molecules consequently do not easily break apart and react with other substances (they have low chemical reactivity) and exhibit special physical characteristics such as π stacking[25][26] (the understanding of which is still the subject of ongoing investigation[27]). In terms of electronic structure, aromaticity describes a conjugated system made of alternating notionally single and double bonds, plus in some cases a lone pair from an occupied p-orbital perpendicular to the plane of the ring. The term "aromatic sextet" for the electronic system which resists disruption (and hence possesses unusual stability) is attributed to Sir Robert Robinson[28] though the idea can be traced further to Crocker[29] and Armstrong.[30] This configuration allows for the delocalization of π electrons around the ring, increasing the molecule's stability. Such molecules cannot be accurately represented by a single structure and are understood to exist as a combination of resonance hybrids, though single representations are frequently chosen as a short-hand for convenience. As an example, the cyclohexatriene structures Kekulé proposed should have alternating single and double bonds of different lengths, yet each is the same and has a length consistent with benzene having six "one-and-a-half" bonds which is logically the superposition of each resonance contributor.[19] A more accurate representation of the electrons following the merging of p orbitals into π-bonds (see illustration above) is Armstrong's inner cycle model as the electron density is evenly distributed around the aromatic ring[11] in molecular orbitals considered to have π symmetry,[31] though this representation is inconvenient in arrow pushing in mechanistic organic chemistry. The quantum mechanical origins of aromaticity were first modelled by Hückel in 1931.[32][33][34]
Characteristics of aromatic systems
An aromatic (or aryl) compound contains a set of covalently bound atoms with specific characteristics:[35]

- A delocalized conjugated π system, most commonly an arrangement of alternating single and double bonds
- Coplanar structure, with all the contributing atoms in the same plane
- Contributing atoms arranged in one or more rings
- A number of π delocalized electrons that is even, but not a multiple of 4. That is, 4n + 2 π-electrons, where n = 0, 1, 2, 3, and so on. This is known as Hückel's rule.
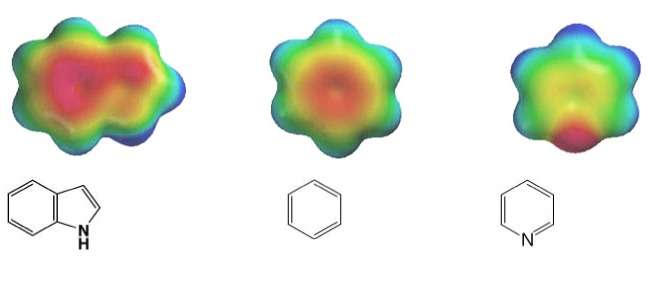
Systems following Hückel's rule, having 4n + 2 π-electrons, are aromatic, but if there are 4n π-electrons along with characteristics 1–3 above, the molecule is said to be antiaromatic. Antiaromatic systems are destabilized and sometimes adopt distorted structures to avoid planarity. An atom in an aromatic system can have other electrons that are not part of the system, and are therefore ignored for the 4n + 2 rule. For example, in furan, the oxygen atom is sp2 hybridized. One lone pair (in the pz orbital) contributes to the π system and the other in the plane of the ring (in the non-bonding sp2 orbital and analogous to the C–H bond in the other positions) does not; consequently, there are 6 π-electrons in furan and it is aromatic. Comparing the nitrogen heterocycles pyrrole and pyridine, each has a 6 π-electron system: the lone pair on the nitrogen atom is required to complete the aromatic sextet in the case of pyrrole while the lone pair of in pyridine is not required. The difference can be seen in the basicity of the two systems, as pyridine can be easily protonated (its lone pair is available for reaction) whilst pyrrole does not react readily.[36] This can be seen in the pKa values of the respective conjugate acids – 8.25 for pyridine compared with −0.27 for pyrrole.[37] In the case of oxazole and isoxazole, the lone pairs contributed to the aromatic sextet come from the occupied pz orbital of oxygen atom rather than the nitrogen lone pairs in hybridized sp2 orbitals. Electrostatic potential surfaces illustrate the difference in electronegativity of the heteroatom bearing an electron pair depending on whether or not it forms part of the aromatic system. In the diagram below, the nitrogen atom in pyridine is markedly more electronegative than nitrogen atom in indole, as indicated by the red regions on the surfaces, because the pyridine nitrogen is free to act as a Lewis base whereas the indole nitrogen is not. The surfaces also illustrate that heteroatoms influence the ability to form cation–pi interactions as cations will be attracted to the aromatic system much more strongly in indole and in benzene than will be the case in pyridine where a direct interaction with the lone pair is more favoured.[38]
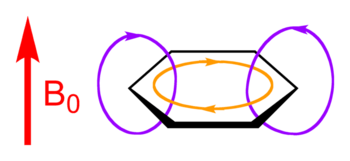
Molecules which can possess an aromatic system will tend to undergo any electronic or conformational structure changes needed to attain aromaticity. Chemical changes which occur as a consequence include a tendency to undergo electrophilic aromatic substitution and nucleophilic aromatic substitution reactions, but not electrophilic addition reactions as happens with carbon–carbon double bonds.[39] This chemical behaviour offers one way to confirm experimentally that an aromatic system is present. The nuclear magnetic resonance (NMR) phenomenon offers a spectroscopic method for demonstrating and investigating aromatic systems as the circulating π-electrons produce ring currents which oppose the applied magnetic field and lead to changes in the measured chemical shifts for both 1H and 13C nuclei.[40][41] X-ray crystal structures confirm the existence of π-π interactions in aromatic systems, such as the dimer of benzene; Perpendicular and offset parallel configurations can be observed in the crystal structures of many simple aromatic compounds.[25][42] More recently, direct measures of electron delocalization allows direct quantification of the extent of aromaticity.[43]
|














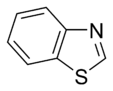
Significance of aromatic systems
Research work undertaken to understand the benzene system opened a new field in organic chemistry leading to the modern understanding of aromatic systems as common throughout the chemical world. Benzene itself is a useful solvent as well as a gasoline additive due to its high octane number and ability to reduce engine knocking, although methylbenzene is now being used as an alternative. Benzene is also a precursor used in the manufacture of styrene, nylon, and epoxy resins. Naphthalene is the active ingredient in traditional mothballs and is used to make the insecticide carbaryl. Aromatic systems appear in many other important industrial products including Bakelite, polystyrene, and PET plastics.[39]
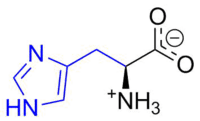
Aromatic systems are common in biological systems. Amongst the 20 standard naturally-occurring amino acids necessary for protein synthesis, four are aromatic – histidine, phenylalanine, tryptophan, and tyrosine.[44] (Histidine is aromatic at all pH levels due to its imidazole side-chain[45] but is also basic and so is often grouped with the two other basic amino acids arginine and lysine.[44]) Aromatic amino acids also serve as the starting materials for biosynthesis of vitamins, neurotransmitters and hormones including dopamine and norepinephrine from tyrosine,[46] serotonin and niacin from tryptophan,[47][48] and tyrosine, thyroxine, and N-methylphenethylamine from phenylalanine.[46][49] Pharmaceuticals with many different applications possess aromatic moieties, as illustrated below.



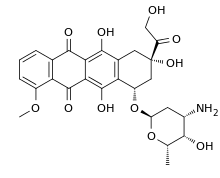
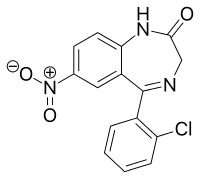


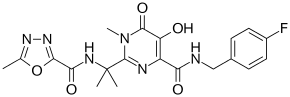


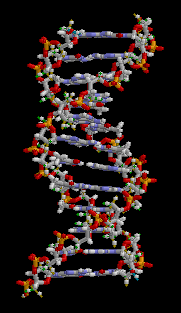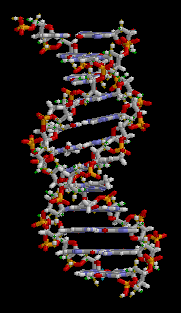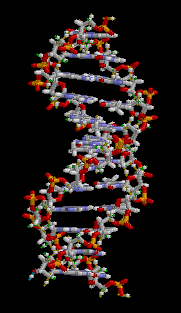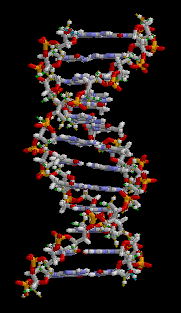
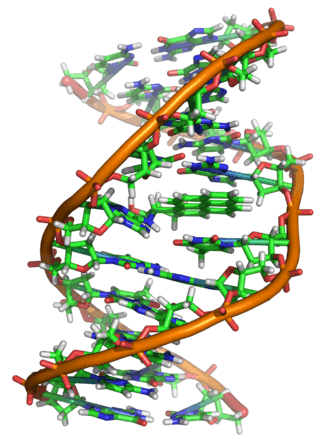
All five of the bases found in DNA and RNA are aromatic – adenine and guanine (purine core structures), and cytosine, thyamine, and uracil (pyrimidine core structures).[62] Biologically active substances including caffeine and uric acid (responsible for gout[63]) also possess purine structures. DNA and RNA have polymeric structures with repeating nucleotide units, each possessing a phosphate group, a ribose or deoxyribose sugar, and a nucleobase. Nucleobases are Watson-Crick base paired through hydrogen bonding,[64] and these pairs are stacked through the center of the double helix through π-π interactions of their aromatic systems.[65] The possibility for extended planar aromatic systems such as acridine (dibenzo[b,e]pyridine) to insert between base base pairs was recognised by Lerman in a process know known as intercalation.[66][67][68] Intercalating agents have applications in biochemical research as stains[69] (ethidium bromide) and in medicine as chemotherapeutic agents (doxorubicin).[55] Polycyclic aromatic hydrocarbons like benzopyrenes and dibenz[a,h]anthracene are some of the 43 known carcinogens in tobacco smoke (the Environment Protection Agency classes tobacco smoke as a class A carcinogen).[39] Benzo[a]pyrene has been shown to form preferentially in lung cancer hotspots[70] and is known to be transformed into a metabolite, (+)-Benzo[a]pyrene-7,8-dihydrodiol-9,10-epoxide, which forms mutagenic adduct with DNA.[71] The epoxide and group on the metabolite alkylate the N2 of guanine bases while the pyrene system intercalates between base pairs, disrupting the structure. Intercalation interferes with DNA replication which can inhibit tumour growth[72] but can also cause changes in the base pair sequence which explains its potential for carcinogeneity.
| ||||||||


-Benzo(a)pyrene-7%2C8-dihydrodiol-9%2C10-epoxide.png)

Formation of new cyclic systems
Alkyne cyclotrimerizations

Many aromatization processes start with two or three acyclic molecules which are fused into a new aromatic ring. Alkyne trimerization is one simple case whereby the [2+2+2] cycloaddition of three alkynes yields a substituted benzene – cyclization of 2-butyne forming hexamethylbenzene, for example.[73] Benzene itself can be prepared by Reppe synthesis with the ligands on the nickel catalyst dictating whether production of benzene or cyclooctatetraene predominates.[1] If an unsymmetric alkyne is used, a variety of products is formed; Rhodium-catalysed cyclotrimerization of phenylacetylene yields both the 1,2,4- and 1,3,5- isomers of triphenylbenzene along with substantial quantities of acyclic enyne products,[74] though use of an alternative catalyst system can produce 97% selectivity for the 1,2,4-product.[75] Antiaromatic cyclobutadiene-derivatives and non-aromatic cyclooctatetraene products are also observed side-products of cyclotrimerizations, and when a mixture of substituted acetylenes is used regioselectivity becomes especially problematic. Replacement of one acetylene by a nitrile allows formation of pyridine or one of its derivatives by Bönnemann cyclization.[76]

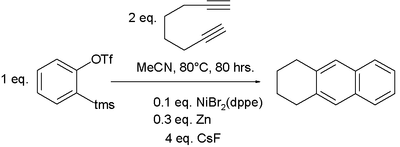
Cyclisation carried out with a diyne and a separate alkyne affords greater control as well as high atom economy. Using commercially available cyclopentadienylcobalt dicarbonyl, CpCo(CO)2, as catalyst, bis(trimethylsilyl)acetylene (BTMSA) will react with a diyne-1,2-disubstituted benzene to form an anthroquinone aromatic system as shown below:[77] Benzyne, generated in situ from a bezene ring bearing ortho-distributed triflate and trimethylsilyl substituents, can be used to generate an aryne in place of an acetylene and combined with a suitable diyne. Such a benzene derivative reacts with 1,7-octadiyne in the presence of a suitable catalyst to generate a naphthalene system.[78] This is an example of a hexadehydro Diels-Alder reaction.

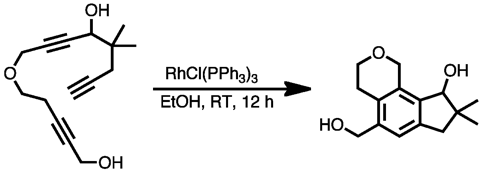
If three alkyne moieties are tethered together, it is possible to create three rings in a single step without the problems with side products mentioned above. An example is seen in the synthesis of calomelanolactone using Wilkinson's catalyst to achieve an intramolecular cyclotrimerization.[79]
Other cyclizations
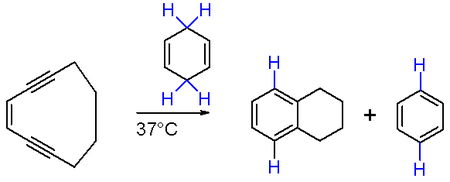
The Bergman cyclization is conceptually similar, using a enediyne with (Z)-stereochemistry plus a hydrogen donor, catalysed by metal centre like ruthenium;[80] (Z)-hex-3-en-1,5-diyne cyclizes to benzene after reduction of the intermediate diradical, for example.[81] The enediyne moiety can be included within an existing ring, allowing access to a bicyclic system under mild conditions as a consequence of the ring strain in the reactant. Cyclodeca-3-en-1,5-diyne reacts with 1,3-cyclohexadiene to produce benzene and tetralin at 37 °C, the reaction being highly favorable owing to the formation of two new aromatic rings:
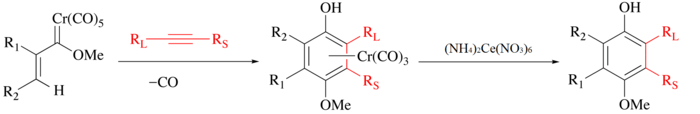
In the Wulff–Dötz reaction an alkyne, carbon monoxide and a chromium carbene complex are the reactants and the product is a chromium half-sandwich complex.[82][83] The phenol ligand can be isolated by mild oxidation with ceric ammonium nitrate.
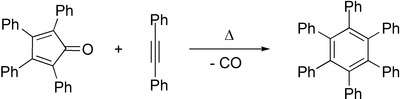
Diels–Alder reactions of alkynes with 2-pyrone[84] or cyclopentadienone with expulsion of carbon dioxide or carbon monoxide, respectively, also form arene compounds. Tetracyclone reacts with diphenylacetylene to form hexaphenylbenzene after loss of carbon monoxide, for example.[85] The Wagner-Jauregg reaction involves a double Diels-Alder addition of maleic anhydride to a 1,1-diarylethylene, resulting in an aryl-substituted naphthalene product. The reaction is unusual in that the aromaticity of the starting material is lost as the aryl-substituted styrene moiety is sufficiently activated to act as a dienophile, but the intermediate undergoes re-aromatization with sulfur.[86]

In a Huisgen cycloaddition, an azide is combined with an alkyne to generate a triazole, and is a specific example of a 1,3-dipolar cycloaddition to a form a heterocyclic five-membered aromatic rings with regio- and stereoselectivity.[87] American chemist Barry Sharpless has described this cycloaddition as "the cream of the crop" of click chemistry[88] and "the premier example of a click reaction."[89]


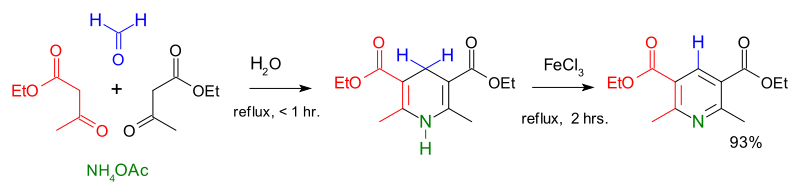
Condensation reactions are another route to the formation of heterocyclic aromatic systems. The Chichibabin pyridine synthesis involves condensation of acrolein (from a Knoevenagel reaction of acetaldehyde with formaldehyde) with acetaldehyde and ammonia, followed by catalysed oxidation of dihydropyridine to pyridine.[90][91] Hantzsch's approach to pyridines dates from 1881 and involves condensation of a β-keto acid or ester with an aldehyde and a nitrogen donor (typicallyammonia or one of its salts.[92] Knoevenagel modified this process for asymmetrically substituted pyridine derivatives.[93] Quinoxaline systems can be formed by the condensation of 1,2-diketones with o-diaminobenzene, quinoxaline itself forming from glyoxal and o-phenylenediamine.[94] Combining benzil with o-phenylenediamine in the presence of 2-iodobenzoic acid (IBX, catalyst) yields 2,3-diphenylquinoxaline in high yield.[95]
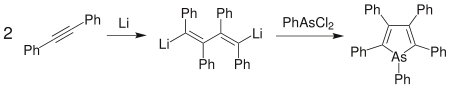
Another effective means of generating heterocyclic systems is to effect ring-closure using the heteroatom. "Moderately aromatic" arsole derivatives can be formed by dimerising diphenylacetylene with lithium to form 1,4-dilithiotetraphenyl-1,3-butadiene.[96] Phenylarsenous dichloride is added to close the ring to form pentaphenylarsole.[97] The diiodo analogue of the lithium salt can be used in its place, and other pnictogen heterocycles can be produced by replacing the arsenic compound as appropriate.
From existing cyclic systems

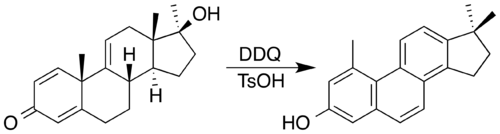
Non aromatic rings can be aromatized through a variety of simple transformations. Dehydration allows the Semmler-Wolff transformation of 2-cyclohexenone oxime to aniline under acidic conditions.[98] In applying redox processes, 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) and an acid catalyst has been used to synthesise a steroid with a phenanthrene core by oxidatation accompanied by a double methyl migration.[99] In the process, DDQ is itself reduced into an aromatic hydroquinone product.
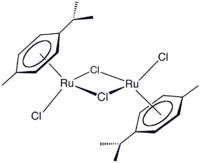
Redox reactions involving transition metals can also be used to produce aromatic systems which then serve as ligands in organometallic complexes. α-Phellandrene (2-methyl-5-iso-propyl-1,3-cyclohexadiene) is oxidised to p-iso-propyltoluene with the reduction of ruthenium trichloride.[100] The resulting ruthenium(II) dimer [(η6-cymene)RuCl2]2] can be cleaved to a monomeric adduct with a suitable Lewis base – [(η6-cymene)RuCl2(PPh3)] is formed with triphenylphosphine, for example. Exchange reactions occur with other arenes and so the p-cymene product can be recovered.

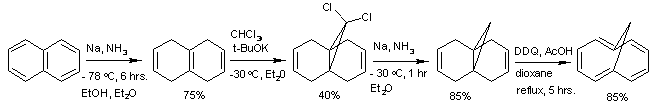
The 10 π-electron system cyclodecapentaene is non-aromatic as it is non-planar, but aromatic derivatives can be prepared. In the following reaction sequence, naphthalene is converted through a series of non-aromatic intermediates to 1,6-methano[10]annulene by Birch reduction, carbene addition, and then DDQ oxidation.[101] Many other conversions of one aromatic system to another are known. One simple example is the industrial synthesis of pyrrole by exchanging the oxygen atom in furan for a nitrogen moiety. Ammonia and solid acid catalysts (like SiO2 and Al2O3) are required.[102] Pyrrole can be converted to 3-chloropyridine by insertion of a carbene into the five-member ring. Dichlorocarbene adds to form a strained bicyclic cyclopropane system which then opens to form the six-member pyridine product, a transformation known as the Ciamician-Dennstedt rearrangement.[103]
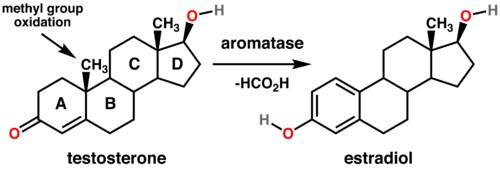
Aromatase is an enzyme which converts some enones with a six membered ring to aromatic rings - specifically testosterone to estradiol and androstenedione to estrone. Each of these aromatizations involves the oxidation of the C-19 methyl group to formic acid to allow for the formation of the aromatic system, conversions which are necessary parts of estrogen tumorogenesis in the development of breast cancer and ovarian cancer in postmenopausal women and gynecomastia in men.[104] Aromatase inhibitors like exemestane (which forms a permanent and deactivating bond with the aromatase enzyme)[105] and anastrozole and letrozole (which compete for the enzyme)[106] have been shown to be more effective than anti-estrogen medications such as tamoxifen likely because they prevent the formation of estradiol.[104]
Ionic systems
Aromaticity can be found in individual ions which satisfy Hückel's rule, which can be accessed by simple redox or acid-base reactions. A Hückel n = 0 cation can be prepared from cyclopropene systems, an example being the bromide salt of the triphenylcycloprenium cation.[107] Ionic Hückel n = 1 systems are known both for anions (cyclopentadienyl) and cations (tropylium). Tropylium, C
7H+
7, was first reported in 1881[108] though its identity was not recognized for over 60 years.[109][110] Tropylium appears regularly in mass spectrometry, given a signal at m/z = 91. Compounds containing the benzyl moiety produce the molecular fragment PhCH+
2, which isomeizes to the stable tropylium cation. Cycloheptatriene (non-aromatic) undergoes a redox reaction with bromine to produce tropylium with the release of hydrogen bromide; it can also be oxidised with chromic acid and a ring-contracting rearrangement occurs in which benzaldehyde is produced:[111]
- C
7H
8 + Br
2 → C
7H+
7 + Br−
+ HBr
- C
7H+
7 + HCrO−
4 → C
6H
5CHO + CrO
2 + H
2O

Cyclopentadiene is more acidic than a typical hydrocarbon (pKa ~ 15-16 compared with cyclopentane's pKa ~ 45) and so can be readily deprotonated to its aromatic conjugate base, an approach used to form compounds like thallium cyclopentadienide,[112] an example of an organometallic polymer.[113] A direct redox reaction of sodium metal with cyclopentadiene yields sodium cyclopentadienide,[114] though a convenient base like sodium hydride can also be used.[115] Compounds like these can be used to prepare metallocene compounds such as ferrocene where two cyclopentadienyl ligands coordinated to a metal centre. Freshly-cracked cyclopentadiene will react with iron(II) chloride and a base to effect deprotonation to produce ferrocene in good yield. Even a weak bases such as amines are sufficient to deprotonate cyclopentadiene for this reaction.[116] Like most aromatic substances, the product here undergoes substitution reactions rather than addition reactions. As an example, Friedel-Crafts acylation of ferrocene with acetic anhydride yields acetylferrocene[117] just as acylation of benzene yields acetophenone under similar conditions.

Corannulene, a [5]circulene, is a fragment of buckminsterfullerene which is found in the buckycatcher shown above and often called a buckybowl due to its bowl-like shape. Motivation for its synthesis was the hypothesis that it would display annulene-within-an-annulene aromaticity, with a 6 π-electron cyclopentadienyl anion core within a 14 π-electron annulenyl cation.[118] Later work has questioned the validity of this model though the molecule is definitely aromatic.[119][120] The dianion has been demonstrated to be antiaromatic and the tetraanion is aromatic.



Cyclooctatetraene has been shown to be non-aromatic[121] and to possess a bent "tub-shaped" conformation with alternating double and single bonds.[122][123] Its planar confirmation would be antiaromatic and is thus disfavoured. By contrast, its dianion C
8H2−
8 is both planar and aromatic and has a Hückel electron count of 10. It is formed readily by direct reaction with potassium metal[124] and uranocene, an analogue of ferrocene, has been prepared from dipotassium cyclooctatetraenide and uranium tetrachloride in tetrahydrofuran at 0 °C:[125]
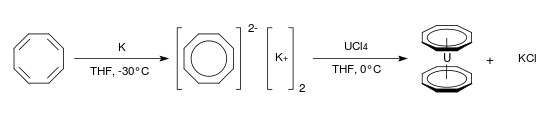
Isomerization
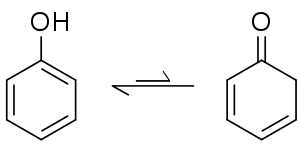

An isomerization reaction involves the rearrangement of atoms within a single molecule to one of its isomers. Some isomerization processes may occur spontaneously, resulting in an equilibrium between the isomers, and the position of the system generally favors an aromatic product if the process is itself an aromatization. Amongst keto-enol tautomers, the keto form is generally favoured on by thermodynamics, but in the case of cyclohexa-2,4-dienone its aromatic isomer phenol is strongly favoured.[126] The equilibrium constant for this system has been determined to be 10−13, meaning that in phenol there are 10 trillion molecules in the aromatic enol form for every molecule in the keto form.[127] Isomerization reactions are temperature dependent, for example melting 1,4-naphthalenediol at 200 °C produces a 2:1 mixture with its keto form, 1,4-dioxotetralin.[128] The conversion to the 1,4-dioxotetralin tautomer can be driven to completion when bound in a chromium tricarbonyl piano stool complex.

A non-tautomeric example of isomerization can be seen in the azulene–napthalene system, a thermal rearrangement[129] in which each of the compounds is stable at room temperature; such interconversion aromatization reactions are rare.[130] Carbon-13 isotope studies have shown scrambling of the α- and β- positions of naphthalene through an azulene intermediate at elevated temperatures[131] though naphthalene has roughly double the aromatic stabilization of azulene. The synthesis of the azulene system is an example of a multi-step organic synthesis, in this case from cycloheptatriene, as shown below.[132] Azulene is unusual in that it is a polar hydrocarbon – the molecular dipole moment of azulene is 1.08 D compared with naphthalene's 0 D.[133] This can be understood by recognizing that one of the important resonance contributors for azulene has the tropylium cation fused to the cyclopentadienyl anion; reactivity studies confirm the electrophilic and nucleophilic characteristics of these rings, respectively.

See also
References
- 1 2 Reppe, W.; Schlichting, O.; Klager, K.; Toepel, T. (1948). "Cyclisierende Polymerisation von Acetylen I Über Cyclooctatetraen". Justus Liebigs Annalen der Chemie (in German). 560: 1–92. doi:10.1002/jlac.19485600102.
- ↑ Morris, E. T. (1984). Fragrance: The Story of Perfume from Cleopatra to Chanel. Charles Scribner's Sons. p. 101. ISBN 0684181959.
- ↑ Faraday, M. (1825). "On New Compounds of Carbon and Hydrogen, and on Certain Other Products Obtained During the Decomposition of Oil by Heat". Philosophical Transactions of the Royal Society. 115: 440–466. doi:10.1098/rstl.1825.0022.
- ↑ Kaiser, R. (1968). "Bicarburet of Hydrogen. Reappraisal of the Discovery of Benzene in 1825 with the Analytical Methods of 1968". Angewandte Chemie International Edition in English. 7 (5): 345–350. doi:10.1002/anie.196803451.
- ↑ Hofmann, A. W. von (1845). "Ueber eine sichere Reaction auf Benzol". Annalen der Chemie und Pharmacie (in German). 55 (2): 200–205. doi:10.1002/jlac.18450550205.
- ↑ Mansfield, C. B. (1849). "Untersuchung des Steinkohlentheers". Annalen der Chemie und Pharmacie (in German). 69 (2): 162–180. doi:10.1002/jlac.18490690203.
- ↑ Hofmann, A. W. von (1856). "On Insolinic Acid". Proceedings of the Royal Society. 8: 1–3. doi:10.1098/rspl.1856.0002.
- ↑ Claus, A. K. L. (1867). "Theoretische Betrachtungen und deren Anwendungen zur Systematik der organischen Chemie". Berichte über die Verhandlungen der Naturforschenden Gesellschaft zu Freiburg im Breisgau (in German). 4: 116–381.
- ↑ Dewar, J. (1867). "On the Oxidation of Phenyl Alcohol, and a Mechanical Arrangement Adapted to Illustrate Structure in the Non-Saturated Hydrocarbons". Proceedings of the Royal Society of Edinburgh. 6: 82–86. doi:10.1017/S0370164600045387.
- ↑ Ladenburg, A. (1869). "Bemerkungen zur aromatischen Theorie". Berichte der Deutschen Chemischen Gesellschaft (in German). 2: 140–142. doi:10.1002/cber.18690020171.
- 1 2 Armstrong, H. E. (1887). "An Explanation of the Laws which Govern Substitution in the case of Benzenoid Compounds". Journal of the Chemical Society. 51: 258–268. doi:10.1039/CT8875100258.
- 1 2 Thiele, J. (1899). "Zur Kenntnis der ungesättigten Verbindungen". Justus Liebigs Annalen der Chemie (in German). 306: 87–142. doi:10.1002/jlac.18993060107.
- ↑ Kekulé, F. A. (1865). "Sur la Constitution des Substances Aromatiques". Bulletin de la Société Chimique de Paris (in French). 3: 98–110.
- ↑ Kekulé, F. A. (1866). "Untersuchungen über Aromatische Verbindungen". Liebigs Annalen der Chemie und Pharmacie (in German). 137 (2): 129–36. doi:10.1002/jlac.18661370202.
- ↑ Read, J. (1995). From Alchemy to Chemistry. New York: Dover Publications. pp. 179–180. ISBN 9780486286907.
- ↑ Rocke, A. J. (2015). "It Began with a Daydream: The 150th Anniversary of the Kekulé Benzene Structure". Angewandte Chemie International Edition in English. 54 (1): 46–50. doi:10.1002/anie.201408034.
- ↑ Lonsdale, K. (1929). "The Structure of the Benzene Ring in C6(CH3)6". Proceedings of the Royal Society A. 123 (792): 494–515. doi:10.1098/rspa.1929.0081.
- ↑ Lonsdale, K. (1931). "An X-Ray Analysis of the Structure of Hexachlorobenzene, Using the Fourier Method". Proceedings of the Royal Society A. 133 (822): 536–553. doi:10.1098/rspa.1931.0166.
- 1 2 "Bonding in Benzene – the Kekulé Structure". www.chemguide.co.uk. Retrieved 31 May 2016.
- ↑ Moran, D.; Simmonett, A. C.; Leach, F. E.; Allen, W. D.; Schleyer, P. V.; Schaefer, H. F. (2006). "Popular Theoretical Methods Predict Benzene and Arenes to be Nonplanar". Journal of the American Chemical Society. 128 (29): 9342–9343. doi:10.1021/ja0630285.
- ↑ Cooper, D. L.; Gerratt, J.; Raimondi, M. (1986). "The Electronic Structure of the Benzene Molecule". Nature. 323 (6090): 699–701. doi:10.1038/323699a0.
- ↑ Pauling, L. (1987). "Electronic Structure of the Benzene Molecule". Nature. 325 (6103): 396. doi:10.1038/325396d0.
- ↑ Sygula, A.; Fronczek, F. R.; Sygula, R.; Rabideau, P. W.; Olmstead, M. M. (2007). "A Double Concave Hydrocarbon Buckycatcher". Journal of the American Chemical Society. 129 (13): 3842–3843. doi:10.1021/ja070616p.
- ↑ McMurry, J. (2007). Organic Chemistry (7th ed.). Brooks-Cole. p. 515. ISBN 0495112585.
- 1 2 Hunter, C. A.; Sanders, J. K. M. (1990). "The Nature of π–π Interactions". Journal of the American Chemical Society. 112 (14): 5525–5534. doi:10.1021/ja00170a016.
- ↑ McGaughey, G. B.; Gagné, M.; Rappé, A. K. (1998). "Pi-Stacking Interactions. Alive and Well in Proteins". Journal of Biological Chemistry. 273 (25): 15458–15463. doi:10.1074/jbc.273.25.15458.
- ↑ Wheeler, S. E.; Bloom, J. W. G. (2014). "Toward a More Complete Understanding of Noncovalent Interactions Involving Aromatic Rings". Journal of Physical Chemistry A. 118 (32): 6133–6147. doi:10.1021/jp504415p.
- ↑ Armit, J. W.; Robinson, R. (1925). "CCXI. Polynuclear Heterocyclic Aromatic Types. Part II. Some Anhydronium Bases". Journal of the Chemical Society, Transactions. 127: 1604–1618. doi:10.1039/CT9252701604.
- ↑ Crocker, E. C. (1922). "Application Of The Octet Theory To Single-Ring Aromatic Compounds". Journal of the American Chemical Society. 44 (8): 1618–1630. doi:10.1021/ja01429a002.
- ↑ Armstrong, H. E. (1890). "The structure of Cycloid Hydrocarbon". Proceedings of the Chemical Society (London). 6 (85): 95–106. doi:10.1039/PL8900600095.
- ↑ Housecroft, C. E.; Sharpe, A. G. (2005). Inorganic Chemistry (2nd ed.). Pearson Prentice Hall. pp. 29–33. ISBN 0130399132.
- ↑ Hückel, E. (1931). "Quantentheoretische Beiträge zum Benzolproblem I. Die Elektronenkonfiguration des Benzols und Verwandter Verbindungen". Zeitschrift für Physik. 70 (3–4): 204–286. doi:10.1007/BF01339530.
- ↑ Hückel, E. (1931). "Quanstentheoretische Beiträge zum Benzolproblem II. Quantentheorie der Induzierten Polaritäten". Zeitschrift für Physik. 72 (5–6): 310–337. doi:10.1007/BF01341953.
- ↑ Hückel, E. (1932). "Quantentheoretische Beiträge zum Problem der Aromatischen und Ungesättigten Verbindungen. III". Zeitschrift für Physik. 76 (9–10): 628–648. doi:10.1007/BF01341936.
- ↑ Ouellette, R. J.; Rawn, J. D. (1996). Organic Chemistry. Prentice Hall. p. 473. ISBN 0023901713.
- ↑ Ghatak, K. L. (2014). "Properties of Molecules II". A Textbook of Organic Chemistry and Problem Analysis. PHI Learning Pvt. Ltd. p. 380. ISBN 9788120347977.
- ↑ Lawrence, S. A. (2004). "An Introduction to Heterocyclic Amines". Amines: Synthesis, Properties and Applications. Cambridge University Press. p. 120. ISBN 9780521782845.
- ↑ Ma, J. C.; Dougherty, D. A. (1997). "The Cation-π Interaction". Chemical Reviews. 97 (5): 1303–1324. doi:10.1021/cr9603744.
- 1 2 3 Seager, S. L.; Slabaugh, M. R. (2013). "Properties and Uses of Aromatic Compounds". Organic and Biochemistry for Today (8th ed.). Cengage Learning. pp. 65–66. ISBN 9781285605906.
- ↑ Merino, G.; Heine, T.; Seifert, G. (2004). "The Induced Magnetic Field in Cyclic Molecules". Chemistry: A European Journal. 10 (17): 4367–4371. doi:10.1002/chem.200400457.
- ↑ Gomes, J. A. N. F.; Mallion, R. B. (2001). "Aromaticity and Ring Currents". Chemical Reviews. 101 (5): 1349–1384. doi:10.1021/cr990323h.
- ↑ Sinnokrot, M. O.; Valeev, E. F.; Sherrill, C. D. (2002). "Estimates of the Ab Initio Limit for π-π Interactions: The Benzene Dimer". Journal of the American Chemical Society. 124 (36): 10887–10893. doi:10.1021/ja025896h.
- ↑ Feixas, F.; Matito, E.; Poater, J.; Solà, M. (2015). "Quantifying Aromaticity with Electron Delocalisation Measures". Chemical Society Reviews. 44 (18): 6434–6451. doi:10.1039/C5CS00066A.
- 1 2 Gropper, S. S.; Smith, J. L. (2012). "Amino Acid Classification". Advanced Nutrition and Human Metabolism (6th ed.). Cengage Learning. pp. 183–186. ISBN 9781133104056.
- ↑ Mrozek, A.; Karolak-Wojciechowska, J.; Kieć-Kononowicz, K. (2003). "Five-Membered Heterocycles. Part III. Aromaticity of 1,3-Imidazole in 5+n Hetero-Bicyclic Molecules". Journal of Molecular Structure. 655 (3): 397–403. Bibcode:2003JMoSt.655..397M. doi:10.1016/S0022-2860(03)00282-5.
- 1 2 Broadley, K. J. (2010). "The Vascular Effects of Trace Amines and Amphetamines". Pharmacology & Therapeutics. 125 (3): 363–375. doi:10.1016/j.pharmthera.2009.11.005.
- ↑ Schaechter, J. D.; Wurtman, R. J. (1990). "Serotonin release varies with brain tryptophan levels". Brain Research. 532 (1-2): 203–210. doi:10.1016/0006-8993(90)91761-5.
- ↑ Ikeda, M.; Tsuji, H.; Nakamura, S; Ichiyama, A.; Nishizuka, Y.; Hayaishi, O. (1965). "Studies on the Biosynthesis of Nicotinamide Adenine Dinucleotide. II. A Role of Picolinic Carboxylase in the Biosynthesis of Nicotinamide Adenine Dinucleotide from Tryptophan in Mammals" (PDF). Journal of Biological Chemistry. 240 (3): 1395–1401. PMID 14284754.
- ↑ Lindemann, L.; Hoener, M. C. (2005). "A Renaissance in Trace Amines Inspired by a Novel GPCR Family". Trends in Pharmacological Sciences. 26 (5): 274–281. doi:10.1016/j.tips.2005.03.007.
- ↑ "Lamotrigine". Drugs.com. Retrieved 1 June 2016.
- ↑ "Tenofovir". Drugs.com. Retrieved 1 June 2016.
- ↑ "Ciprofloxacin". Drugs.com. Retrieved 1 June 2016.
- ↑ "Oxprenolol". Drugs.com. Retrieved 1 June 2016.
- ↑ "Codeine". Drugs.com. Retrieved 1 June 2016.
- 1 2 "Doxorubicin". Drugs.com. Retrieved 1 June 2016.
- ↑ "Clonazepam". Drugs.com. Retrieved 1 June 2016.
- ↑ "Lornoxicam". Drugs.com. Retrieved 12 June 2016.
- ↑ Spiller, H. A.; Winter, M. L.; Weber, J. A.; Krenzelok, E. P.; Anderson, D. L.; Ryan, M. L. (2003). "Skin Breakdown and Blisters from Senna-Containing Laxatives in Young Children". Annals of Pharmacotherapy. 37 (5): 636–639. doi:10.1345/aph.1C439.
- ↑ "Raltegravir". Drugs.com. Retrieved 12 June 2016.
- ↑ "Chloroquine". Drugs.com. Retrieved 1 June 2016.
- ↑ "Fluoxetine Hydrochloride". Drugs.com. Retrieved 1 June 2016.
- ↑ Verma, S.; Eckstein, F. (1998). "Modified Oligonucleotides: Synthesis and Strategy for Users". Annual Review of Biochemistry. 67: 99–134. doi:10.1146/annurev.biochem.67.1.99.
- ↑ Chen, L. X.; Schumacher, H. R. (2008). "Gout: an Evidence-Based Review". Journal of Clinical Rheumatology. 14 (5): S55–62. doi:10.1097/RHU.0b013e3181896921.
- ↑ Watson, J. D.; Crick, F. (1953). "A Structure for Deoxyribose Nucleic Acid". Nature. 171 (4356): 737–738. doi:10.1038/171737a0.
- ↑ Protozanova, E.; Yakovchuk, P.; Frank-Kamenetskii, M. D. (2004). "Stacked–Unstacked Equilibrium at the Nick Site of DNA". Journal of Molecular Biology. 342 (3): 775–785. doi:10.1016/j.jmb.2004.07.075.
- ↑ Lerman, L. S. (1961). "Structural Considerations in the Interaction of DNA and Acridines". Journal of Molecular Biology. 3 (1): 18–30. doi:10.1016/S0022-2836(61)80004-1.
- ↑ Luzzati, V. F.; Lerman, L. S. (1961). "Interaction of DNA and Proflavine: A Small-Angle X-Ray Scattering Study". Journal of Molecular Biology. 3 (5): 634–639. doi:10.1016/S0022-2836(61)80026-0.
- ↑ Lerman, L. S. (1963). "The Structure of the DNA-Acridine Complex". Proceedings of the National Academy of Sciences of the United States of America. 49 (1): 94–102. doi:10.1073/pnas.49.1.94.
- ↑ Sabnis, R. W. (2010). Handbook of Biological Dyes and Stains: Synthesis and Industrial Application. Wiley. ISBN 9780470407530.
- ↑ Denissenko, M. F.; Pao, A.; Tang, M.-S.; Pfeifer, G. P. (1996). "Preferential Formation of Benzo[a]pyrene Adducts at Lung Cancer Mutational Hotspots in P53". Science. 274 (5286): 430–432. doi:10.1126/science.274.5286.430.
- 1 2 Pradhan, P.; Tirumala, S.; Liu, X.; Sayer, J. M.; Jerina, D. M.; Yeh, H. J. C. (2001). "Solution Structure of a Trans-Opened (10S)-dA Adduct of (+)-(7S,8R,9S,10R)-7,8-Dihydroxy-9,10-epoxy-7,8,9,10-tetrahydrobenzo[a]pyrene in a fully Complementary DNA Duplex: Evidence for a Major Syn Conformation". Biochemistry. 40 (20): 5870–5881. doi:10.1021/bi002896q. PMID 11352722.
- ↑ Fornari, F. A.; Randolph, J. K.; Yalowich, J. C.; Ritke, M. K.; Gewirtz, D. A. (1994). "Interference by Doxorubicin with DNA Unwinding in MCF-7 Breast Tumor Cells". Molecular Pharmacology. 45 (4): 649–656. PMID 8183243.
- ↑ Weber, S. R.; Brintzinger, H. H. (1977). "Reactions of Bis(hexamethylbenzene)iron(0) with Carbon Monoxide and with Unsaturated Hydrocarbons". Journal of Organometallic Chemistry. 127 (1): 45–54. doi:10.1016/S0022-328X(00)84196-0.
- ↑ Ardizzoia, G. A.; Brenna, S.; Cenini, S.; LaMonica, G.; Masciocchi, N.; Maspero, A. (2003). "Oligomerization and Polymerization of Alkynes Catalyzed by Rhodium(I) Pyrazolate Complexes". Journal of Molecular Catalysis A: Chemical. 204–205: 333–340. doi:10.1016/S1381-1169(03)00315-7.
- ↑ Hilt, G.; Vogler, T.; Hess, W.; Galbiati, F. (2005). "A Simple Cobalt Catalyst System for the Efficient and Regioselective Cyclotrimerisation of Alkynes". Chemical Communications. 2005 (11): 1474–1475. doi:10.1039/b417832g.
- ↑ Behr, A. (2008). Angewandte homogene Katalyse (in German). Wiley-VCH. ISBN 9783527316663.
- ↑ Hillard, R. L.; Vollhardt, K. P. C. (1977). "Substituted Benzocyclobutenes, Indans, and Tetralins via Cobalt-Catalyzed Cooligomerization of α,ω-diynes with Substituted Acetylenes. Formation and Synthetic Utility of Trimethylsilylated Benzocycloalkenes". Journal of the American Chemical Society. 99 (12): 4058–4069. doi:10.1021/ja00454a026.
- ↑ Hsieh, J.-C.; Cheng, C.-H. (2005). "Nickel-Catalyzed Cocyclotrimerization of Arynes with Diynes; A Novel Method for Synthesis of Naphthalene Derivatives". Chemical Communications. 2005 (19): 2459–2461. doi:10.1039/b415691a.
- ↑ Neeson, S. J.; Stevenson, P. J. (1988). "Rhodium Catalysed [2+2+2] Cycloadditions – An Efficient Regiospecific Route to Calomelanolactone". Tetrahedron Letters. 29 (7): 813–814. doi:10.1016/S0040-4039(00)80217-8.
- ↑ Odedra, A.; Wu, C.-J.; Pratap, T. B.; Huang, C.-W.; Ran, Y.-F.; Liu, R.-S. (2005). "Ruthenium-Catalyzed Aromatization of Enediynes via Highly Regioselective Nucleophilic Additions on a π-Alkyne Functionality. A Useful Method for the Synthesis of Functionalized Benzene Derivatives". Journal of the American Chemical Society. 127 (10): 3406–3412. doi:10.1021/ja043047j.
- ↑ Mohamed, R. K.; Peterson, P. W.; Alabugin, I. V. (2013). "Concerted Reactions that Produce Diradicals and Zwitterions: Electronic, Steric, Conformational and Kinetic Control of Cycloaromatization Processes". Chemical Reviews. 113 (9): 7089–7129. doi:10.1021/cr4000682.
- ↑ Waters, M.; Wulff, W. D. (2008). "The Synthesis of Phenols and Quinones via Fischer Carbene Complexes". Organic Reactions. 70 (2): 121–623. doi:10.1002/0471264180.or070.02.
- ↑ Dötz, K. H. (1983). "Carbon-Carbon Bond Formation via Carbonyl-Carbene Complexes". Pure and Applied Chemistry. 55 (11). doi:10.1351/pac198355111689.
- ↑ Woodard, B. T.; Posner, G. H. (1999). "Recent Advances in Diels-Alder Cycloadditions Using 2-Pyrones". In Harmata, M. Advances in Cycloaddition. 5. JAI Press. pp. 47–83. doi:10.1002/chin.199935304. ISBN 9780762303465.
- ↑ Fieser, L. F. (1966). "Hexaphenylbenzene". Org. Synth. 46: 44.; Coll. Vol., 5, p. 604 doi:10.15227/orgsyn.046.0044
- ↑ Bergmann, F.; Szmuszkowicz, J.; Fawaz, G. (1947). "The Condensation of 1,1-Diarylethylenes with Maleic Anhydride". Journal of the American Chemical Society. 69 (7): 1773–1777. doi:10.1021/ja01199a055.
- ↑ Huisgen, Rolf (1963). "1.3-Dipolare Cycloadditionen Rückschau und Ausblick". Angewandte Chemie (in German). 75: 604–637. doi:10.1002/ange.19630751304.
- ↑ Kolb, H. C.; Finn, M. G.; Sharpless, K. B. (2001). "Click Chemistry: Diverse Chemical Function from a Few Good Reactions". Angewandte Chemie International Edition. 40 (11): 2004–2021. doi:10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5.
- ↑ Kolb, H. C.; Sharpless, B. K. (2003). "The Growing Impact of Click Chemistry on Drug Discovery". Drug Discovery Today. 8 (24): 1128–1137. doi:10.1016/S1359-6446(03)02933-7.
- ↑ Frank, R.L.; Seven, R. P. (1949). "Pyridines. IV. A Study of the Chichibabin Synthesis". Journal of the American Chemical Society. 71 (8): 2629–2635. doi:10.1021/ja01176a008.
- ↑ Shimizu, S.; Watanabe, N.; Kataoka, T.; Shoji, T.; Abe, N.; Morishita, S.; Ichimura, H. (2005). "Pyridine and Pyridine Derivatives". Ullmann's Encyclopedia of Industrial Chemistry. Wiley-VCH. doi:10.1002/14356007.a22_399.
- ↑ Hantzsch, A. (1881). "Condensationsprodukte aus Aldehydammoniak und ketonartigen Verbindungen". Berichte der Deutschen Chemischen Gesellschaft (in German). 14 (2): 1637–1638. doi:10.1002/cber.18810140214.
- ↑ Knoevenagel, E.; Fries, A. (1898). "Synthesen in der Pyridinreihe. Ueber eine Erweiterung der Hantzsch'schen Dihydropyridinsynthese". Berichte der Deutschen Chemischen Gesellschaft (in German). 31 (1): 761–767. doi:10.1002/cber.189803101157.
- ↑ Jones, R. G.; McLaughlin, K. C. (1950). "2,3-Pyrazinedicarboxylic Acid". Organic Syntheses. 30: 86. doi:10.15227/orgsyn.030.0086.
- ↑ Heravi, M. M.; Bakhtiari, K.; Tehrani, M. H.; Javadi, N. M.; Oskooie, H. A. (2006). "Facile Synthesis of Quinoxaline Derivatives Using o-Iodoxybenzoic Acid (IBX) at Room Temperature". Arkivoc. 2006 (16): 16–22. doi:10.3998/ark.5550190.0007.g02.
- ↑ Johansson, M. P.; Juselius, J. (2005). "Arsole Aromaticity Revisited". Letters in Organic Chemistry. 2: 469–474. doi:10.2174/1570178054405968.
Using quantum chemical methodology, we reinvestigate the aromaticity of the much debated arsole, using the newly developed gauge-including magnetically induced currents (GIMIC) method. GIMIC provides a quantitative measure of the induced ring current strength, showing arsole to be moderately aromatic.
- ↑ Braye, E. H.; Hübel, W.; Caplier, I. (1961). "New Unsaturated Heterocyclic Systems. I". Journal of the American Chemical Society. 83 (21): 4406–4413. doi:10.1021/ja01482a026.
- ↑ Horning, E. C.; Stromberg, V. L.; Lloyd, H. A. (1952). "Beckmann Rearrangements. An Investigation of Special Cases". Journal of the American Chemical Society. 74 (20): 5153–5155. doi:10.1021/ja01140a048.
- ↑ Brown, W.; Turner, A. B. (1971). "Applications of High-Potential Quinones. Part VII. The Synthesis of Steroidal Phenanthrenes by Double Methyl Migration". Journal of the Chemical Society C: Organic: 2566–2572. doi:10.1039/J39710002566.
- ↑ Bennett, M. A.; Huang, T. N.; Matheson, T. W.; Smith, A. K. (1982). "(η6-Hexamethylbenzene)ruthenium Complexes". Inorganic Syntheses. 21: 74–78.
- ↑ Vogel, E.; Klug, W.; Breuer, A. (1974). "1,6-Methano[10]annulene". Organic Synthesis. 54: 11. doi:10.15227/orgsyn.054.0011.
- ↑ Harreus, A. L. (2002). "Pyrrole". Ullmann's Encyclopedia of Industrial Chemistry. Wiley-VCH. doi:10.1002/14356007.a22_453.
- ↑ Skell, P. S.; Sandler, S. R. (1958). "Reactions of 1,1-Digalocyclopropanes with Electrophilic Reagents. Synthetic Route for Inserting a Carbon Atom Between the Atoms of a Double Bond". Journal of the American Chemical Society. 80 (8): 2024–2025. doi:10.1021/ja01541a070.
- 1 2 Avendaño, C.; Menéndez, J. C. (2008). "Aromatase Inhibitors". Medicinal Chemistry of Anticancer Drugs. Elsevier. pp. 65–73. doi:10.1016/B978-0-444-52824-7.00003-2. ISBN 9780080559629.
- ↑ Jasek, W., ed. (2007). Austria-Codex (in German) (62nd ed.). Vienna: Österreichischer Apothekerverlag. pp. 656–660. ISBN 9783852001814.
- ↑ Dinnendahl, V.; Fricke, U., eds. (2007). Arzneistoff-Profile (in German). 4 (21st ed.). Eschborn, Germany: Govi Pharmazeutischer Verlag. ISBN 9783774198463.
- ↑ Xu, R.; Breslow, R. (1997). "1,2,3-Triphenylcyclopropenium Bromide". Organic Syntheses. 74: 72. doi:10.15227/orgsyn.074.0072.
- ↑ Merling, G. (1891). "Ueber Tropin". Berichte der Deutschen Chemischen Gesellschaft (in German). 24 (2): 3108–3126. doi:10.1002/cber.189102402151.
- ↑ Doering, W. von E.; Knox, L. H. (1954). "The Cycloheptatrienylium (Tropylium) Ion". Journal of the American Chemical Society. 76 (12): 3203–3206. doi:10.1021/ja01641a027.
- ↑ Balaban, A. T.; Oniciu, D. C.; Katritzky, A. R. (2004). "Aromaticity as a Cornerstone of Heterocyclic Chemistry". Chemical Reviews. 104 (5): 2777–2812. doi:10.1021/cr0306790.
- ↑ Agarwai, O. P. (2009). Reactions and Reagents (46th ed.). Krishna Prakashan Media. pp. 614–615. ISBN 9788187224655.
- ↑ Nielson, A. J.; Rickard, C. E. F.; Smith, J. M. (1986). "Cyclopentadienylthallium (Thallium Cyclopentadienide)". Inorganic Syntheses. 24: 97–99. doi:10.1002/9780470132555.ch31.
- ↑ Olbrich, F.; Behrens, U. (1997). "Crystal Structure of Catena-Cyclopentadienyl Thallium, [Tl(C5H5)]". Zeitschrift für Kristallographie – New Crystal Structures. 212 (1): 47–47. doi:10.1524/ncrs.1997.212.1.47.
- ↑ Cotton, F. A.; Wilkinson, G. (1999). Advanced Inorganic Chemistry (6th ed.). John Wiley and Sons. ISBN 9780471199571.
- ↑ Girolami, G. S.; Rauchfuss, T. B.; Angelici, R. J. (1999). Synthesis and Technique in Inorganic Chemistry. University Science Books. ISBN 0935702482.
- ↑ Wilkinson, G. (1956). "Ferrocene". Organic Syntheses. 36: 31. doi:10.15227/orgsyn.036.0031.
- ↑ Bozak, R. E. (1966). "Acetylation of Ferrocene: A Chromatography Experiment for Elementary Organic Laboratory". Journal of Chemical Education. 43 (2): 73. doi:10.1021/ed043p73.
- ↑ Barth, W. E.; Lawton, R. G. (1966). "Dibenzo[ghi,mno]fluoranthene". Journal of the American Chemical Society. 88 (2): 380–381. doi:10.1021/ja00954a049.
- ↑ Sygula, A.; Rabideau, P. W. (1995). "Structure and Inversion Barriers of Corannulene, its Dianion and Tetraanion. An ab initio Study". Journal of Molecular Structure: THEOCHEM. 333 (3): 215–226. doi:10.1016/0166-1280(94)03961-J.
- ↑ Monaco, G.; Scott, L.; Zanasi, R. (2008). "Magnetic Euripi in Corannulene". The Journal of Physical Chemistry A. 112 (35): 8136–8147. doi:10.1021/jp8038779.
- ↑ Johnson, A. W. (1947). "Organic Chemistry". Science Progress. 35 (139): 506–515. JSTOR 43413011.
- ↑ Kaufman, H. S.; Fankuchen, I.; Mark, H. (1948). "Structure of Cyclo-octatetraene". Nature. 161 (4083): 165. doi:10.1038/161165a0.
- ↑ Thomas, P. M.; Weber, A. (1978). "High Resolution Raman Spectroscopy of Gases with Laser Sources. XIII – The Pure Rotational Spectra of 1,3,5,7-Cyclooctatetraene and 1,5-Cyclooctadiene". Journal of Raman Spectroscopy. 7 (6): 353–357. doi:10.1002/jrs.1250070614.
- ↑ Katz, T. J. (1960). "The Cyclooctatetraenyl Dianion". Journal of the American Chemical Society. 82 (14): 3784–3785. doi:10.1021/ja01499a077.
- ↑ Streitwieser, A.; Mueller-Westerhoff, U. (1968). "Bis(cyclooctatetraenyl)uranium (Uranocene). A New Class of Sandwich Complexes that Utilize Atomic f Orbitals". Journal of the American Chemical Society. 90 (26): 7364–7364. doi:10.1021/ja01028a044.
- ↑ Clayden, J.; Greeves, N.; Warren, S.; Wothers, P. (2001). Organic Chemistry (1st ed.). Oxford University Press. p. 531. ISBN 9780198503460.
- ↑ Capponi, M.; Gut, I. G.; Hellrung, B.; Persy, G.; Wirz, J. (1999). "Ketonization Equilibria of Phenol in Aqueous Solution". Canadian Journal of Chemistry. 77 (5-6): 605–613. doi:10.1139/cjc-77-5-6-605.
- ↑ Kündig, E. P.; Garcia, A. E.; Lomberget, T.; Bernardinelli, G. (2005). "Rediscovery, Isolation, and Asymmetric Reduction of 1,2,3,4-Tetrahydronaphthalene-1,4-dione and Studies of its [Cr(CO)3] Complex". Angewandte Chemie International Edition. 45 (1): 98–101. doi:10.1002/anie.200502588.
- ↑ Scott, L. T. (1982). "Thermal Rearrangements of Aromatic Compounds". Accounts of Chemical Research. 15 (2): 52–58. doi:10.1021/ar00074a004.
- ↑ Scott, L. T.; Kirms, M. A. (1981). "Azulene Thermal Rearrangements. 13C–Labeling Studies of Automerization and Isomerization to Naphthalene". Journal of the American Chemical Society. 103 (19): 5875–5879. doi:10.1021/ja00409a042.
- ↑ Scott, L. T.; Agopian, G. K. (1977). "Automerization of Naphthalene". Journal of the American Chemical Society. 99 (13): 4506–4507. doi:10.1021/ja00455a053.
- ↑ Carret, S.; Blanc, A.; Coquerel, Y.; Berthod, M.; Greene, A. E.; Deprés, J.-P. (2005). "Approach to the Blues: A Highly Flexible Route to the Azulenes". Angewandte Chemie International Edition. 44 (32): 5130–5133. doi:10.1002/anie.200501276.
- ↑ Anderson, A. G.; Stecker, B. M. (1959). "Azulene. VIII. A Study of the Visible Absorption Spectra and Dipole Moments of Some 1- and 1,3-Substituted Azulenes". Journal of the American Chemical Society. 81 (18): 4941–4946. doi:10.1021/ja01527a046.