Peutz–Jeghers syndrome
| Peutz-Jeghers syndrome | |
|---|---|
 | |
| Micrograph of Peutz-Jeghers type colonic polyp. H&E stain. | |
| Classification and external resources | |
| Specialty | medical genetics |
| ICD-10 | Q85.8 |
| ICD-9-CM | 759.6 |
| OMIM | 175200 |
| DiseasesDB | 9905 |
| MedlinePlus | 000244 |
| eMedicine | med/1807 article/182006article/1664349 |
| MeSH | D010580 |
| GeneReviews | |
| Orphanet | 2869 |
Peutz–Jeghers syndrome (often abbreviated PJS) is an autosomal dominant genetic disorder characterized by the development of benign hamartomatous polyps in the gastrointestinal tract and hyperpigmented macules on the lips and oral mucosa (melanosis).[1] This syndrome can be classed as one of various hereditary intestinal polyposis syndromes[2] and one of various hamartomatous polyposis syndromes.[3] It has an incidence of approximately 1 in 25,000 to 300,000 births.[4]
Diagnosis
The main criteria for clinical diagnosis are:
- Family history
- Mucocutaneous lesions causing patches of hyperpigmentation in the mouth and on the hands and feet. The oral pigmentations are the first on the body to appear, and thus play an important part in early diagnosis. Intraorally, they are most frequently seen on the gingiva, hard palate and inside of the cheek. The mucosa of the lower lip is almost invariably involved as well.
- Hamartomatous polyps in the gastrointestinal tract. These are benign polyps with an extraordinarily low potential for malignancy.
Having 2 of the 3 listed clinical criteria indicates a positive diagnosis. The oral findings are consistent with other conditions, such as Addison's disease and McCune-Albright syndrome, and these should be included in the differential diagnosis. 90-100% of patients with a clinical diagnosis of PJS have a mutation in the STK11/LKB1 gene. Molecular genetic testing for this mutation is available clinically.[5]
Prognosis
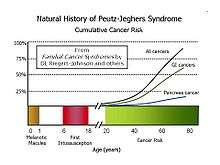
Most patients will develop flat, brownish spots (melanotic macules) on the skin, especially on the lips and oral mucosa, during the first year of life, and a patient’s first bowel obstruction due to intussusception usually occurs between the ages of six and 18 years. The cumulative lifetime cancer risk begins to rise in middle age. Cumulative risks by age 70 for all cancers, gastrointestinal (GI) cancers, and pancreatic cancer are 85%, 57%, and 11%, respectively.
A 2011 Dutch study followed 133 patients for 14 years. The cumulative risk for cancer was 40% and 76% at ages 40 and 70, respectively. 42 (32%) of the patients died during the study, of which 28 (67%) were cancer related. They died at a median age of 45. Mortality was increased compared with the general population.[6]
Genetics
In 1998, a gene was found to be associated with the mutation. On chromosome 19, the gene known as STK11 (LKB1)[7] is a possible tumor suppressor gene. It is inherited in an autosomal dominant pattern, which means that anyone who has PJS has a 50% chance of passing the disease on to their offspring.
Limited evidence base
Peutz–Jeghers syndrome is rare and studies typically include only a small number of patients. Even in those few studies that do contain a large number of patients, the quality of the evidence is limited due to pooling patients from many centers, selection bias (only patients with health problems coming from treatment are included), and historical bias (the patients reported are from a time before advances in the diagnosis of treatment of Peutz–Jeghers syndrome were made). Probably due to this limited evidence base, cancer risk estimates for Peutz–Jeghers syndrome vary from study to study.[8]
Presentation
The risks associated with this syndrome include a strong tendency of developing cancer in a number of parts of the body.[9] While the hamartomatous polyps themselves only have a small malignant potential (<3% - OHCM), patients with the syndrome have an increased risk of developing carcinomas of the pancreas,[10] liver, lungs, breast, ovaries, uterus, testicles and other organs.
The average age of first diagnosis is 23, but the lesions can be identified at birth by an astute pediatrician. Prior to puberty, the mucocutaneous lesions can be found on the palms and soles. Often the first presentation is as a bowel obstruction from an intussusception which is a common cause of mortality; an intussusception is a telescoping of one loop of bowel into another segment.
Cancer screening
Some suggestions for surveillance for cancer include the following:
- Small intestine with small bowel radiography every 2 years,
- Esophagogastroduodenoscopy and colonoscopy every 2 years,
- CT scan or MRI of the pancreas yearly,
- Ultrasound of the pelvis (women) and testes (men) yearly,
- Mammography (women) from age 25 annually livelong,[5] and
- Papanicolaou smear (Pap smear) every year
Follow-up care should be supervised by a physician familiar with Peutz–Jeghers syndrome. Genetic consultation and counseling as well as urological and gynecological consultations are often needed.
Treatment
Resection of the polyps is required only if serious bleeding or intussusception occurs. Enterotomy is performed for removing large, single nodules. Short lengths of heavily involved intestinal segments can be resected. Colonoscopy can be used to snare the polyps if they are within reach.
Eponym
First described in a published case report in 1921 by Johannes Laurentius Augustinus Peutz (1886-1957), a Dutch Internist, it was later formalized into the syndrome by an American internist, Harold Joseph Jeghers (1904-1990) in 1949. [11]
See also
References
- ↑ James, William; Berger, Timothy; Elston, Dirk (2005). Andrews' Diseases of the Skin: Clinical Dermatology (10th ed.). Saunders. p. 857. ISBN 0-7216-2921-0.
- ↑ Dean, PA (1996), "Hereditary intestinal polyposis syndromes", Rev Gastroenterol Mex, 61 (2): 100–111, PMID 8927912.
- ↑ Jelsig, AM; et al. (2014), "Hamartomatous polyposis syndromes: a review", Orphanet J Rare Dis, 15 (9): 101–111, doi:10.1186/1750-1172-9-101, PMC 4112971
 , PMID 25022750.
, PMID 25022750. - ↑ Bouquot, Jerry E.; Neville, Brad W.; Damm, Douglas D.; Allen, Carl P. (2008). Oral and Maxillofacial Pathology. Philadelphia: Saunders. p. 16.11. ISBN 1-4160-3435-8.
- 1 2 McGarrity, Thomas J; Amos, Christopher I; Frazier, Marsha L; Wei, Chongjuan (July 25, 2013). "Peutz-Jeghers Syndrome". In Pagon, Roberta A; Adam, Margaret P; Bird, Thomas D; Dolan, Cynthia R; Fong, Chin-To; Smith, Richard JH; Stephens, Karen. GeneReviews. Seattle: University of Washington. PMID 20301443.
- ↑ van Lier, Margot G. F.; Westerman, Anne Marie; Wagner, Anja; Looman, Caspar W. N.; Wilson, J. H. Paul; de Rooij, Felix W. M.; Lemmens, Valery E. P. P.; Kuipers, Ernst J.; Mathus-Vliegen, Elisabeth M. H. (2011-02-01). "High cancer risk and increased mortality in patients with Peutz-Jeghers syndrome". Gut. 60 (2): 141–147. doi:10.1136/gut.2010.223750. ISSN 1468-3288. PMID 21205875.
- ↑ Universal protein resource accession number Q15831 for "Serine/threonine-protein kinase STK11" at UniProt.
- ↑ Riegert-Johnson, Douglas; Gleeson, Ferga C.; Westra, Wytske; Hefferon, Timothy; Song, Louis M. Wong Kee; Spurck, Lauren; Boardman, Lisa A. (August 9, 2008). "Peutz-Jeghers Syndrome". In Riegert-Johnson, Douglas L; Boardman, Lisa A; Hefferon, Timothy; Roberts, Maegan. Cancer Syndromes.
- ↑ Boardman, Lisa A.; Thibodeau, Stephen N.; Schaid, Daniel J.; Lindor, Noralane M.; McDonnell, Shannon K.; Burgart, Lawrence J.; Ahlquist, David A.; Podratz, Karl C.; Pittelkow, Mark; Hartmann, Lynn C. (1998). "Increased Risk for Cancer in Patients with the Peutz-Jeghers Syndrome". Annals of Internal Medicine. 128 (11): 896–9. doi:10.7326/0003-4819-128-11-199806010-00004. PMID 9634427.
- ↑ Ryan DP, Hong TS, Bardeesy N (September 2014). "Pancreatic adenocarcinoma". N. Engl. J. Med. 371 (11): 1039–49. doi:10.1056/NEJMra1404198. PMID 25207767.
- ↑ Familial Cancer 2002; 1:181-185)
External links
| Wikimedia Commons has media related to Peutz–Jeghers syndrome. |
- GeneReviews/NCBI/NIH/UW entry on Peutz-Jeghers syndrome
- Peutz-Jeghers syndrome - Genetics Home Reference