Lhermitte–Duclos disease
| Lhermitte-Duclos disease | |
|---|---|
 | |
| Classification and external resources | |
| OMIM | 158350 |
| MeSH | D006223 |
Lhermitte–Duclos disease (LDD) (English /ˌlɛrˈmiːtˌduːˈkloʊ/), also called dysplastic gangliocytoma of the cerebellum, is a rare, slowly growing tumor of the cerebellum, a gangliocytoma sometimes considered to be a hamartoma, characterized by diffuse hypertrophy of the granular layer of the cerebellum. It is often associated with Cowden syndrome.[1] It was described by Jacques Jean Lhermitte and P. Duclos in 1920.[2]
Signs and symptoms
Main clinical signs and symptoms include:
Patients with Lhermitte–Duclos disease and Cowden's syndrome may also have multiple growths on skin. The tumor, though benign, may cause neurological injury including abnormal movements.
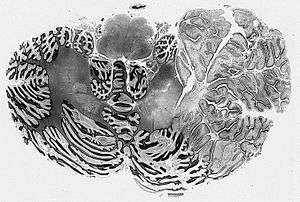
MICROSCOPY(lhermitte-duclos disease) 1>Enlarged circumscribed cerebellar folia 2>internal granular layer is focally indistinct and is occupied by large ganglion cells 3>myelinated tracks in outer molecular layer 4>underlying white matter is atrophic and gliotic
Pathophysiology
In Lhermitte–Duclos disease, the cerebellar cortex loses its normal architecture, and forms a hamartoma in the cerebellar hemispheres. The tumors are usually found on the left cerebellar hemisphere, and consist of abnormal hypertrophic ganglion cells that are somewhat similar to Purkinje cells. The amount of white matter in the cerebellum is diminished.
Like cowden syndrome, patients with Lhermitte–Duclos disease often have mutations in enzymes involved in the Akt/PKB signaling pathway, which plays a role in cell growth. Mutation in PTEN gene on chromosome no. 10q leads to increased activity of AKT and mTOR pathways.
Treatment
Treatment is not needed in the asymptomatic patient. Symptomatic patients may benefit from surgical debulking of the tumor. Complete tumor removal is not usually needed and can be difficult due to the tumor location.
Epidemiology
Lhermitte–Duclos disease is a rare entity; approximately 222 cases of LDD have been reported in medical literature.[3] Symptoms of the disease most commonly manifest in the third and fourth decades of life, although it may onset at any age.[4] Men and women are equally affected, and there is not any apparent geographical pattern.[4]
History
The disease was first described in 1920 by Lhermitte and Duclos.[4]
See also
References
- ↑ Eng C (November 2000). "Will the real Cowden syndrome please stand up: revised diagnostic criteria". J. Med. Genet. 37 (11): 828–30. doi:10.1136/jmg.37.11.828. PMC 1734465
 . PMID 11073535.
. PMID 11073535. - ↑ Lhermitte J, Duclos P (1920). "Sur un ganglioneurome diffuse du cortex du cervelet". Bulletin de l'Association Francaise pour l'etude du Cancer. Paris. 9: 99–107.
- ↑ Robinson S, Cohen AR (2006). "Cowden disease and Lhermitte-Duclos disease: an update. Case report and review of the literature". Neurosurg Focus. 20 (1): E6. doi:10.3171/foc.2006.20.1.7. PMID 16459996.
- 1 2 3 Kumar, R; Vaid, VK; Kalra, SK (July 2007). "Lhermitte-Duclos disease.". Child's Nervous System. 23 (7): 729–32. doi:10.1007/s00381-006-0271-8. PMID 17221273.
External links
- Lhermitte-Duclos syndrome at Who Named It?
- MedPix: Lhermitte-Duclos — Radiology and Pathology