Thyrotoxic periodic paralysis
| Thyrotoxic periodic paralysis | |
|---|---|
 | |
| Thyrotoxic periodic paralysis occurs when the thyroid gland releases excessive amounts of thyroxine (thyroid hormone). | |
| Classification and external resources | |
| Specialty | neurology |
| ICD-10 | G72.3 |
| ICD-9-CM | 359.3 |
| OMIM | 188580 613239 |
| DiseasesDB | 29122 |
| MedlinePlus | 000319 |
| eMedicine | article/1171678 |
| MeSH | D020514 |
| GeneReviews | |
Thyrotoxic periodic paralysis (TPP) is a condition featuring attacks of muscle weakness in the presence of hyperthyroidism (overactivity of the thyroid gland). Hypokalemia (a decreased potassium level in the blood) is usually present during attacks. The condition may be life-threatening if weakness of the breathing muscles leads to respiratory failure, or if the low potassium levels lead to cardiac arrhythmias (irregularities in the heart rate).[1][2] If untreated, it is typically recurrent in nature.[1]
The condition has been linked with genetic mutations in genes that code for certain ion channels that transport electrolytes (sodium and potassium) across cell membranes. The main ones are the L-type calcium channel α1-subunit[1] and potassium inward rectifier 2.6;[3] it is therefore classified as a channelopathy.[3] The abnormality in the channel is thought to lead to shifts of potassium into cells, under conditions of high thyroxine (thyroid hormone) levels, usually with an additional precipitant.
Treatment of the hypokalemia, followed by correction of the hyperthyroidism, leads to complete resolution of the attacks. It occurs predominantly in males of Chinese, Japanese, Vietnamese, Filipino, and Korean descent.[1] TPP is one of several conditions that can cause periodic paralysis.[4]
Signs and symptoms
An attack often begins with muscle pain, cramping, and stiffness.[5] This is followed by weakness or paralysis that tends to develop rapidly, usually in late evening or the early hours of the morning. The weakness is usually symmetrical;[5] the limb muscles closer to the trunk (proximal) are predominantly affected, and weakness tends to start in the legs and spread to the arms. Muscles of the mouth and throat, eyes, and breathing are usually not affected, but occasionally weakness of the respiratory muscles can cause life-threatening respiratory failure. Attacks typically resolve within several hours to several days, even in the absence of treatment.[1][2][5] On neurological examination during an attack, flaccid weakness of the limbs is noted; reflexes are usually diminished, but the sensory system is unaffected.[1][5] Mental status is not affected.[5]
Attacks may be brought on by physical exertion, drinking alcohol, or eating food high in carbohydrates or salt. This may explain why attacks are more common in summer, when more people drink sugary drinks and engage in exercise. Exercise-related attacks tend to occur during a period of rest immediately after exercise; exercise may therefore be recommended to abort an attack.[1]
There may be symptoms of thyroid overactivity, such as weight loss, a fast heart rate, tremor, and perspiration;[1][2] but such symptoms occur in only half of all cases.[5] The most common type of hyperthyroidism, Graves' disease, may additionally cause eye problems (Graves' ophthalmopathy) and skin changes of the legs (pretibial myxedema).[6] Thyroid disease may also cause muscle weakness in the form of thyrotoxic myopathy, but this is constant rather than episodic.[5]
Causes
Genetics
Genetic mutations in the L-type calcium channel α1-subunit (Cav1.1) have been described in Southern Chinese with TPP. The mutations are located in a different part of the gene from those described in the related condition familial periodic paralysis. In TPP, the mutations described are single-nucleotide polymorphisms located in the hormone response element responsive to thyroid hormone, implying that transcription of the gene and production of ion channels may be altered by increased thyroid hormone levels. Furthermore, mutations have been reported in the genes coding for potassium voltage-gated channel, Shaw-related subfamily, member 4 (Kv3.4) and sodium channel protein type 4 subunit alpha (Na41.4).[1]
Of people with TPP, 33% from various populations were demonstrated to have mutations in KCNJ18, the gene coding for Kir2.6, an inward-rectifier potassium ion channel. This gene, too, harbors a thyroid response element.[3]
Certain forms of human leukocyte antigen (HLA)—especially B46, DR9, DQB1*0303, A2, Bw22, AW19, B17, and DRW8—are more common in TPP. Linkage to particular forms of HLA, which plays a central role in the immune response, might imply an immune system cause, but it is uncertain whether this directly causes TPP or whether it increases the susceptibility to Graves' disease, a known autoimmune disease.[1]
Thyroid disease
The most common underlying form of thyroid disease associated with TPP is Graves' disease, a syndrome due to an autoimmune reaction that leads to overproduction of thyroid hormone.[6] TPP has also been described in people with other thyroid problems such as thyroiditis, toxic nodular goiter, toxic adenoma, TSH-producing pituitary adenoma, excessive ingestion of thyroxine or iodine,[1] and amiodarone-induced hyperthyroidism.[2]
Mechanism
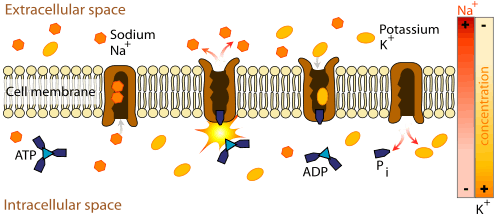
The muscle weakness and increased risk of irregular heart beat in TPP result from markedly reduced levels of potassium in the bloodstream. Potassium is not in fact lost from the body, but increased Na+/K+-ATPase activity (the enzyme that moves potassium into cells and keeps sodium in the blood) leads to shift of potassium into tissues, and depletes the circulation. In other types of potassium derangement, the acid-base balance is usually disturbed, with metabolic alkalosis and metabolic acidosis often being present. In TPP, these disturbances are generally absent. Hypokalemia leads to hyperpolarization of muscle cells, making the neuromuscular junction less responsive to normal nerve impulses and leading to decreased contractility of the muscles.[1]
It is not clear how the described genetic defects increase the Na+/K+-ATPase activity, but it is suspected that the enzyme becomes more active due to increased thyroid hormone levels. Hyperthyroidism increases the levels of catecholamines (such as adrenaline) in the blood, increasing Na+/K+-ATPase activity.[5] The enzyme activity is then increased further by the precipitating causes. For instance, increased carbohydrate intake leads to increased insulin levels; this is known to activate Na+/K+-ATPase. Once the precipitant is removed, the enzyme activity returns to normal levels.[1] It has been postulated that male hormones increase Na+/K+-ATPase activity, and that this explains why males are at a higher risk of TPP despite thyroid disease being more common in females.[2]
TPP is regarded as a model for related conditions, known as "channelopathies", which have been linked with mutations in ion channels; the majority of these conditions occurs episodically.[3]
Diagnosis
Hypokalemia (low blood potassium levels) commonly occurs during attacks; levels below 3.0 mmol/l are typically encountered. Magnesium and phosphate levels are often found to be decreased. Creatine kinase levels are elevated in two thirds of cases, usually due to a degree of muscle injury; severe elevations suggestive of rhabdomyolysis (muscle tissue destruction) are rare.[1][2] Electrocardiography (ECG/EKG) may show tachycardia (a fast heart rate) due to the thyroid disease, abnormalities due to cardiac arrhythmia (atrial fibrillation, ventricular tachycardia), and conduction changes associated with hypokalemia (U waves, QRS widening, QT prolongation, and T wave flattening).[2] Electromyography shows changes similar to those encountered in myopathies (muscle diseases), with a reduced amplitude of the compound muscle action potentials (CMAPs);[4] they resolve when treatment has commenced.[1]
TPP is distinguished from other forms of periodic paralysis (especially hypokalemic periodic paralysis) with thyroid function tests on the blood. These are normal in the other forms, and in thyrotoxicosis the levels of thyroxine and triiodothyronine are elevated, with resultant suppression of TSH production by the pituitary gland.[1][6] Various other investigations are usually performed to separate the different causes of hyperthyroidism.[6]
Treatment
In the acute phase of an attack, administration of potassium will quickly restore muscle strength and prevent complications. However, caution is advised as the total amount of potassium in the body is not decreased, and it is possible for potassium levels to overshoot ("rebound hyperkalemia"); slow infusions of potassium chloride are therefore recommended while other treatment is commenced.[1]
The effects of excess thyroid hormone typically respond to the administration of a non-selective beta blocker, such as propranolol (as most of the symptoms are driven by increased levels of adrenaline and its effect on the β-adrenergic receptors). Subsequent attacks may be prevented by avoiding known precipitants, such as high salt or carbohydrate intake, until the thyroid disease has been adequately treated.[1]
Treatment of the thyroid disease usually leads to resolution of the paralytic attacks. Depending on the nature of the disease, the treatment may consist of thyrostatics (drugs that reduce production of thyroid hormone), radioiodine, or occasionally thyroid surgery.[1][2]
Epidemiology
TPP occurs predominantly in males of Chinese, Japanese, Vietnamese, Filipino, and Korean descent,[1] as well as Thais,[3] with much lower rates in people of other ethnicities.[1] In Chinese and Japanese people with hyperthyroidism, 1.8–1.9% experience TPP. This is in contrast to North America, where studies report a rate of 0.1–0.2%.[1][2] Native Americans, who share a genetic background with East Asians, are at an increased risk.[1]
The typical age of onset is 20–40. It is unknown why males are predominantly affected, with rates in males being 17- to 70-fold those in females, despite thyroid overactivity being much more common in women.[1][2]
History

After several case reports in the 18th and 19th centuries, periodic paralysis was first described in full by the German neurologist Carl Friedrich Otto Westphal (1833–1890) in 1885.[7][8] In 1926 the Japanese physician Tetsushiro Shinosaki, from Fukuoka, observed the high rate of thyroid disease in Japanese people with periodic paralysis.[9][10] The first English-language report, in 1931, originated from Dunlap and Kepler, physicians at the Mayo Clinic; they described the condition in a patient with features of Graves' disease.[2][10] In 1937 periodic paralysis was linked with hypokalemia, as well as precipitation of attacks with glucose and insulin.[11][12] This phenomenon has been used as a diagnostic test.[12]
In 1974 it was discovered that propranolol could prevent attacks.[13] The concept of channelopathies and the link with specific ion channel mutations emerged at the end of the 20th century.[1][3][4]
References
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 Kung AW (July 2006). "Clinical review: Thyrotoxic periodic paralysis: a diagnostic challenge". J. Clin. Endocrinol. Metab. 91 (7): 2490–5. doi:10.1210/jc.2006-0356. PMID 16608889.
- 1 2 3 4 5 6 7 8 9 10 11 Pothiwala P, Levine SN (2010). "Analytic review: thyrotoxic periodic paralysis: a review". J. Intensive Care Med. 25 (2): 71–7. doi:10.1177/0885066609358849. PMID 20089526.
- 1 2 3 4 5 6 Ryan DP, Ptácek LJ (October 2010). "Episodic neurological channelopathies". Neuron. 68 (2): 282–92. doi:10.1016/j.neuron.2010.10.008. PMID 20955935.
- 1 2 3 Fontaine B (2008). "Periodic paralysis". Adv. Genet. Advances in Genetics. 63: 3–23. doi:10.1016/S0065-2660(08)01001-8. ISBN 978-0-12-374527-9. PMID 19185183.
- 1 2 3 4 5 6 7 8 Lin SH (January 2005). "Thyrotoxic periodic paralysis" (PDF). Mayo Clin. Proc. 80 (1): 99–105. doi:10.4065/80.1.99. PMID 15667036.
- 1 2 3 4 Weetman AP (October 2000). "Graves' disease". N. Engl. J. Med. 343 (17): 1236–48. doi:10.1056/NEJM200010263431707. PMID 11071676.
- ↑ Westphal CF (1885). "Über einen merkwürdigen Fall von periodischer Lähmung aller vier Extremitäten mit gleichzeitigem Erlöschen der elektrischen Erregbarkeit während der Lähmung". Berl. Klin. Wochenschr. (in German). 22: 489–91 and 509–11.
- ↑ Sternberg D, Tabti N, Hainque B, Fontaine B (28 April 2009). "Hypokalemic Periodic Paralysis". GeneReviews. PMID 20301512.
- ↑ Shinosaki T (1926). "Klinische Studien über die periodische Extremitätenlähmung". Z. Gesamt. Neurol. Psychiatr. (in German). 100 (1): 564–611. doi:10.1007/BF02970940.
- 1 2 Dunlap H, Kepler K (1931). "A syndrome resembling familial periodic paralysis occurring in the course of exophthalmic goiter". Endocrinology. 15 (6): 541–6. doi:10.1210/endo-15-6-541.
- ↑ Aitken RS, Allott EN, Castleden LI, Walker M (1937). "Observations on a case of familial periodic paralysis". Clin. Sci. 3: 47–57.
- 1 2 McFadzean AJ, Yeung R (February 1967). "Periodic paralysis complicating thyrotoxicosis in Chinese". Br Med J. 1 (5538): 451–5. doi:10.1136/bmj.1.5538.451. PMC 1840834
 . PMID 6017520.
. PMID 6017520. - ↑ Yeung RT, Tse TF (October 1974). "Thyrotoxic periodic paralysis. Effect of propranolol". Am. J. Med. 57 (4): 584–90. doi:10.1016/0002-9343(74)90010-2. PMID 4432863.