Pathology of multiple sclerosis
Multiple sclerosis can be pathologically defined as the presence of distributed glial scars (sclerosis or scleroses, in its plural form) in the central nervous system that must show dissemination in time (DIT) and in space (DIS) to be considered MS lesions.[1][2]
The scars that give the name to the condition are produced by the astrocyte cells healing old lesions.[3] These glial scars are the remainings of previous demyelinating inflammatory lesions (encephalomyelitis disseminata) which are produced by one or more unknown underlying conditions.
Apart of the disseminated lesions that define the condition, the CNS white matter shows normally other kind of damages. At least five characteristics are present in CNS tissues of MS patients: Inflammation beyond classical white matter lesions (NAWM, NAGM), intrathecal Ig production with oligoclonal bands, an environment fostering immune cell persistence, Follicle-like aggregates in the meninges (B-cells mostly infected with EBV[4]) and a disruption of the blood–brain barrier even outside of active lesions.[5]
Confluent subpial cortical lesions are the most specific finding for MS, being exclusively present in MS patients.[6] Though this feature can only be detected during an autopsy[7] there are some subrogate markers under study[8] Damage in MS consists also in areas with hidden damage (normal appearing white and gray matters) and two kinds of cortical lesions: Neuronal loss and cortical demyelinating lesions. The neural loss is the result of neural degeneration from lesions located in the white matter areas and the cortical demyelinating lesions are related to meningeal inflammation.[9][10]
The scars in the white matter are known to appear from confluence of smaller ones[11]
Currently the term "multiple sclerosis" is ambiguous and refers not only to the presence of the scars, but also to the unknown underlying condition that produces these scars. Besides clinical diagnosis uses also the term "multiple sclerosis" for speaking about the related clinical courses. Therefore, when referring to the presence of the scars is better to use the equivalent term Astrocytic fibrillary gliosis[9]
Lesions consistent with MS

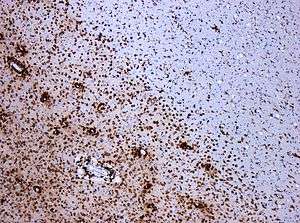
A combination of histologic and/or immunohistochemical stains can be used to visualize post-mortem MS characteristic lesions and to diagnose post-mortem "inflammatory demyelinating lesions consistent with MS":[12]
- hematoxylin and eosin stain (demonstrates tissue and cell morphology)
- myelin stains (Luxol fast blue/periodic acid-Schiff, Luxol fast blue/hematoxylin/eosin, or immunohistochemistry for myelin proteins)
- macrophage-specific markers (immunohistochemistry for KiM1P or CD68)
- stains for axons (Bielschowsky silver impregnation or immunohistochemistry for neurofilament protein)
- stains for astrocytes (hematoxylin and eosin or immunohistochemistry for glial fibrillary acidic protein) and
- stains for the different lymphocyte subtypes (immunohistochemistry for CD3, CD4, CD8, CD20, and/or CD138)
These markers are specific for the different processes that drive the formation of plaques: inflammation, myelin breakdown, astrogliosis, oligodendrocyte injury, neurodegeneration and axonal loss, and remyelination. MS lesions evolve differently during early versus chronic disease phases, and within each phase, different kind of activity appears.
Nevertheless, to consider the lesions representative of MS they also must be disseminated in time and space, being the dissemination in time shown by the stage of the lesion evolution.
Demyelination process
Though lesions in MS are heterogeneous and there are four different ways in which they start, they share many things in common. Specially important in the development process are some white matter areas, which are abnormal under MRI, named NAWM (normal appearing white matter) because its where the lesions appear.
Damage occurs in two phases. First some MRI-abnormal areas with hidden damage appear in the brain and spine (NAWM, NAGM, DAWM), followed later by leaks in the blood–brain barrier where immune cells infiltrate causing the known demyelination.[13]
In the most common pattern of MS, a special subset of lymphocytes, called T helper cells, specifically those releasing Th2 cytokines, play a key role in the development of the lesion.[14][15] The functional characterization shows that T cells releasing Th2 cytokines and helping B cells dominate the T-cell infiltrate in pattern II brain lesions.[14] Also Th1 and Th17 could be implicated[16]
A protein called Interleukin 12 is responsible for the differentiation of naive T cells into inflammatory T cells. An over production of this protein is what causes the increased inflammation in MS patients.[17] Under normal circumstances, these lymphocytes can distinguish between self and non-self. However, in a person with MS, these cells recognize healthy parts of the central nervous system as foreign and attack them as if they were an invading virus, triggering inflammatory processes and stimulating other immune cells and soluble factors like cytokines and antibodies. Many of the myelin-recognizing T cells belong to a terminally differentiated subset called co-stimulation-independent effector-memory T cells.[18][19][20][21][22][23][24][25][26][27][28]
Recently other type of immune cells, B Cells, have been also implicated in the pathogenesis of MS[29] and in the degeneration of the axons.[30] and the oligodendrocytes.[31]
The axons themselves can also be damaged by the attacks.[32] Often, the brain is able to compensate for some of this damage, due to an ability called neuroplasticity. MS symptoms develop as the cumulative result of multiple lesions in the brain and spinal cord. This is why symptoms can vary greatly between different individuals, depending on where their lesions occur.
Repair processes, called remyelination, also play an important role in MS. Remyelination is one of the reasons why, especially in early phases of the disease, symptoms tend to decrease or disappear temporarily. Nevertheless, nerve damage and irreversible loss of neurons occur early in MS.
The oligodendrocytes that originally formed a myelin sheath cannot completely rebuild a destroyed myelin sheath. However, the central nervous system can recruit oligodendrocyte stem cells capable of proliferation and migration and differentiation into mature myelinating oligodendrocytes. The newly formed myelin sheaths are thinner and often not as effective as the original ones. Repeated attacks lead to successively fewer effective remyelinations, until a scar-like plaque is built up around the damaged axons. These scars are the so-called "scleroses" that define the condition. They are named glial scars because they are produced by glial cells, mainly astrocytes, and their presence prevents remyelination. Therefore, there is research ongoing to prevent their formation.
Under laboratory conditions, stem cells are quite capable of proliferating and differentiating into remyelinating oligodendrocytes; it is therefore suspected that inflammatory conditions or axonal damage somehow inhibit stem cell proliferation and differentiation in affected areas[33]
Specific areas of damage
The unknown underlying condition produces inflammation, demyelination and atrophy in sereval areas. Notice that some of the listed areas, like the retina, do not have myelin. In these cases only inflammation and atrophy appears.
Brain lesions distribution

Multiple sclerosis is considered a disease of the white matter because normally lesions appear in this area, but it is also possible to find some of them in the grey matter.[34]
Using high field MRI system, with several variants several areas show lesions, and can be spacially classified in infratentorial, callosal, juxtacortical, periventricular, and other white matter areas.[35] Other authors simplify this in three regions: intracortical, mixed gray-white matter, and juxtacortical.[36] Others classify them as hippocampal, cortical, and WM lesions,[37] and finally, others give seven areas: intracortical, mixed white matter-gray matter, juxtacortical, deep gray matter, periventricular white matter, deep white matter, and infratentorial lesions.[38] The distribution of the lesions could be linked to the clinical evolution[39]
Post-mortem autopsy reveal that gray matter demyelination occurs in the motor cortex, cingulate gyrus, cerebellum, thalamus and spinal cord.[40] Cortical lesions have been observed specially in people with SPMS but they also appear in RRMS and clinically isolated syndrome. They are more frequent in men than in women[41] and they can partly explain cognitive deficits.
Regarding two parameters of the cortical lesions, fractional anisotropy (FA) is lower and mean diffusivity (MD) is higher in patients than in controls.[42] The differences are larger in SPMS (secondary progressive multiple sclerosis) than in RRMS (relapsing-remitting multiple sclerosis) and most of them remain unchanged for short follow-up periods. They do not spread into the subcortical white matter and never show gadolinium enhancement. Over a one-year period, CLs can increase their number and size in a relevant proportion of MS patients, without spreading into the subcortical white matter or showing inflammatory features similar to those of white matter lesions.[43]
Due to the distribution of the lesions, since 1916 they are also known as Dawson's fingers.[44] They appear around the brain blood vessels.
Spinal cord damage

Cervical spinal cord has been found to be affected by MS even without attacks, and damage correlates with disability.[45] In RRMS, cervical spinal cord activity is enhanced, to compensate for the damage of other tissues.[46] It has been shown that Fractional anisotropy of cervical spinal cord is lower than normal, showing that there is damage hidden from normal MRI.[47]
Progressive tissue loss and injury occur in the cervical cord of MS patients. These two components of cord damage are not interrelated, suggesting that a multiparametric MRI approach is needed to get estimates of such a damage. MS cord pathology is independent of brain changes, develops at different rates according to disease phenotype, and is associated to medium-term disability accrual.[48]
Spinal cord presents grey matter lesions, that can be confirmed post-mortem and by high field MR imaging. Spinal cord grey matter lesions may be detected on MRI more readily than GM lesions in the brain, making the cord a promising site to study the grey matter demyelination.[49] Myelin Water Fraction (MWF) shows lesions under MRI[50]
Cerebellum and Thalamus
Cerebellar ataxia appears mainly in PPMS and it is related to the pathological changes in the cerebellum. Some special cells present only in the cerebellum, Purkinje cells, have been reported to be part of this problems. Increasing of neurofilament phosphorylation has been reported[51]
Cerebellum is specially affected in progressive variants. Grey matter damage in the cerebellum is linked to inflammation in the subarachnoid space[52] Though most of the cerebellum damage occurs in late stages, it can be seen that there are abnormalities since early disease stages[53] mostly of the "Normal Appearing" kind[54]
Thalamus degeneration in MS presents several features, such as trans-neuronal or Wallerian degeneration.[55]
Cortex
Around 26% of MS lesions appear inside or adjacent to the cortex.[56] It seems that in RRMS patients, both deep and cortical GM atrophy are associated with pathology in connected white matter.[57] Cortical lesions are inflammatory (immune mediated) and can present relapses[58]
Cortex lesions are disposed around the principal cortical veins and the majority enter the terrain of the white matter, and have been classified into seven types[56]
Some research groups have proposed that cortical lesions are the origin of the NAWM areas in the white matter[59] and 7 Tesla scanners seem to confirm this hypothesis, showing that cortical pathology starts in the pial surface (external layer of the brain), which is in contact with the CSF, and extends later into the brain inner layers[60]
Lesions in the cortex have been classified by the area they affect into four groups: type I (leukocortical), type II (intracortical), type III (subpial), and type IV (subpial extending through the whole cortical width but not to subcortical WM). This classification is not related to the white matter lesions classification.[61][62]
Normal appearing cortex
As with Normal appearing white matter (NAGM) and gray matter (NAGM), there is a normal appearing cortex in which no lesions have developed, but with abnormal microscopical properties.
Recently it has been found that Normal Appearing Cortex presents primary neurodegenerative damage in the dendritic spines of the neurons, with no demyelination nor autoimmune infiltrates. For some authors this constitutes a proof to state that MS is a primary neurodegenerative condition.[63]
Motor cortex
fibrinogen is deposited in MS motor cortex and associates with neurodegeneration.[64]
Olfactory bulb
The olfactory nerve, similar to the optic nerve, is part of the Central Nervous System. This nerve terminates in the olfactory bulb, which also belongs to the central nervous system. Both develop from the CNS embrion, and recently it has been shown, by autopsies, that they are affected by the same diseases than the rest of the CNS.[65] In particular, they are damaged during the multiple sclerosis course.
Related to this, the CSF of patients with disease activity show high levels of "Lateral Olfactory Tract Usher Substance" (LOTUS)[66]
Retina and optic nerve damage
The eye's retina in MS is also damage. Given that retina cells have no myelin, damage must be different from the autoimmune attack of the brain. The underlying condition in the retina produces pure neurodegeneration.[67]
The retina and the optic nerve originate as outgrowths of the brain during embryonic development, so they are considered part of the central nervous system (CNS).[68] It is the only part of the CNS that can be imaged non-invasively in the living organism. The retina nerve fiber layer (RNFL) is thinner than normal in MS patients[69]
The procedure by which the MS underlying condition attacks the retina is currently unknown, but seems mediated by human leukocyte antigen-DR positive cells with the phenotype of microglia.[70]
MS patients show axonal loss in the retina and optic nerve, which can be measured by Optical coherence tomography[71] or by Scanning laser polarimetry.[72] This measure can be used to predict disease activity[73] and to establish a differential diagnosis from Neuromyelitis optica[74]
About antibodies in the retina, tissue-bound IgG was demonstrated on retinal ganglion cells in six of seven multiple sclerosis cases but not in controls.[75] Two eye problems, Uveitis and retinal phlebitis are manifestations of MS.[76]
Proposed procedures for the neurodegeneration are than Narrower arterioles and wider venules have been reported.[77] Also rigidity has been noticed[78]
Degenerative process in the optic nerve and retina
Human retina is devoid of myelin, but inflammation is prominent in MS even at late stages of disease, showing prominent gliosis and inflammation surrounding the vessels of the inner retina.[79]
Some results suggest the presence of trans-synaptic degeneration as a contributor to chronic axon damage in the optic nerve and retina[80] Nevertheless, the authors of the paper were unable to identify whether the degeneration condition spreads from the anterior part or from the rear.
The optic radiation (OR), which is a set of axons that lead to the visual cortex, is more similar to the rest of the brain because it contains myelin. It is also damaged. In this area NAWM areas (see below) appear. The optic radiation damage is composed by two factors: trans-synaptic degeneration, and wallerian degeneration [81]
Respect the theory about the role of the meninges in MS evolution, it is important to notice that the optic nerve in its intraorbital part has the tree meninges and it is tightly coupled with the pia mater.[82]
Neural and axonal damage
Two different mechanisms of axon destruction are acting in MS. First of all, there is a diffuse axon degeneration, probably related to the NAWM appearance. Later, there is a second axonal damage mechanism localized in old demyelinating lesions, probably produced by B-Cells. This second damage is related to the T1-Hypointense lesions (MRI black holes) which appear when a demyelinating lesion is not remyelinated.[83]
The axons of the neurons are damaged probably by B-Cells,[30] though currently no relationship has been established with the relapses or the attacks.[32] It seems that this damage is a primary target of the immune system, i.e. not secondary damage after attacks against myelin,[84] though this has been disputed[85]
Proton magnetic resonance spectroscopy has shown that there is widespread neuronal loss even at the onset of MS, largely unrelated to inflammation.[86]
A relationship between neural damage and N-Acetyl-Aspartate concentration has been established, and this could lead to new methods for early MS diagnostic through magnetic resonance spectroscopy[87]
Axonal degeneration at CNS can be estimated by N-acetylaspartate to creatine (NAA/Cr) ratio, both measured by with proton magnetic resonance spectroscopy.[88]
The meninges in multiple sclerosis
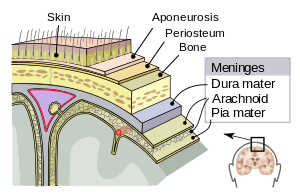
The meninges are three layers that protect the brain and the spinal cord. They are called (from the outside to the inside) the dura mater, the arachnoid mater and the pia mater. The cerebrospinal fluid flows between the second and the third one. A remarkable finding in MS is that some Follicle-like aggregates appear in the meninges (composed by B-cells mostly infected with EBV[89]). These aggregates grow during the disease process and is mostly found in secondary progressive patients.
Inflammation in the meninges has been found to be associated to gray mater (cortical) demyelination. Besides subpial demyelination suggest either a problem in the CSF or in the pia mater that should protect the cortex[90]
Whatever the underlying condition for MS is, some damage is triggered by a CSF unknown soluble factor, which is produced in meningeal areas and diffuses into the cortical parenchyma. It destroys myelin either directly or indirectly through microglia activation.[6]
The infiltration into meninges, which has been referred to as Tertiary Lymphoid Tissues (TLTs), prepares the infiltration into the CNS parenchyma causing demyelination in subpial and cortical areas. Animal models suggest that infiltrating Th17 cells remodel the meningeal stromal (non-immune) cells and initiate the formation of TLTs during EAE. The remodeled stromal cells retain and promote the production of Th17 and the accumulation of B cells. The collaboration between LTB on Th17 cells and LTBR (Lymphotoxin beta receptor) on meningeal radio-resistant cells is very crucial for the induction and progression of MS.[91]
Peripheral nervous system involvement
Though MS is defined as a CNS condition, some reports link problems in the peripheral nervous system with the presence of MS plaques in the CNS[92]
Lesion structure and evolution

MS lesions mainly consist in demyelination and scarring in the fatty myelin sheaths around the axons of the brain and spinal cord.[93]
Lesions evolve from the Normal Appearing White Matter. In MTR-MRI, the apparent diffusion coefficient (ADCav) is a measure of water molecule motion. It can be seen that before the BBB breakdown, this coeficient increases until, at some point, the blood-brain barrier breaks down and immune cells enter the brain producing the lesion.[94]
According with the most recent research, an active lesion is composed of different layers:[95]
- NAWM border with the lesion: These areas contained activated microglia, antibodies binding to astrocytes, axons, oligodendrocytes and dendritic cells along blood vessels. No T or B cells are present.
- Lesion external layer: Number of oligodendrocyte cell bodies decreases. Remaining oligodendrocytes are sometimes swollen or dying. Myelin sheaths are still intact but swollen. Small increase in microglia and T cells.
- Active layer: Phagocytic demyelinating areas: There is myelin debris taken up by local microglia and phagocytes entering from the bloodstream. More T cells in these areas, and in the space adjacent to blood vessels.
- Recently demyelinated tissue: Tissues were full of myelin-containing phagocytes. Signs of early remyelination together with small numbers of oligodendrocytes. Large numbers of T cells, B cells, and other immune cells concentrated around blood vessels.
- Inactive layer: Again activated microglia and dendritic cells were also found around blood vessels.
Some lesions named "slowly eroding lesions" or "slowly expanding" feature myelin phagocytosis at the lesion edge and evolve expanding across the white matter[96]
Lesions under MRI
Most MS lesions are isointense to white matter (they appear bright) on T1-weighted MRI, but some are "hypointense" (lower intensity). These are called "black holes" (BH). They appear specially in the supratentorial region of the brain.
When BH's appear, around half of them revert in a month. This is considered a sign of remyelination. When they remain, this is regarded as a sign of permanent demyelination and axonal loss. This has been shown on post-mortem autopsies.[97]
Small lesions are invisible under MRI. Therefore, clinically assisted diagnostic criteria are still required for a more accurate MS diagnosis than MRI alone.[98]
The lesion evolution under MRI has been reported to begin as a pattern of central hyperintensity. This was seen in the majority of new lesions, both on proton density and contrast-enhanced T1-weighted images.[99] When gadolinium is used, the lesion expansion can be classified as nodular or ringlike[100]
Whatever the demyelination process is, currently it is possible to detect lesions before demyelination, and they show clusters of activated microglia and leukocyte infiltration, together with oligodendrocytes abnormalities.[101] Some research groups consider some areas of the NAWM with clusters of microglial nodules as "preactive MS lesions".[102]
Lesion evolution can be followed via MRI[103]
Damage before BBB disruption
Special MRI methods
The classic MRI methods are named T1-relaxation and T2-relaxation. They create the images based in the "relaxation time", i.e., the time it takes for a molecule to realign its magnetic with its environment after an electromagnetic pulse has taken it out of the equilibrium.
A third type of MRI is based in the water diffusivity. It is called "Diffusion MRI" or "Diffusion Tensor MRI". and the images produced are normally named Diffusion Tensor Images (DTI). A modification of the image post-processing is to account for the water density in each area. These are called "Diffusion Weighted Images" (DWI) or Difussion Tensor MRI, DT-MRI. The diffusion measures the water response and the tensor structure takes account of the orientation of the tissue fibers. It is important because NAWM and NAGM show abnormal DT-MRI[104]
A fourth important MRI technique is the Magnetization Transfer technique, MT-MRI. It measures differences in the Magnetization Transfer Ration (MTR). The idea is that the nucleus of any atom that has a net nuclear spin and that is bonded to a hydrogen atom could potentially be imaged via "heteronuclear magnetization transfer MRI". This would image the high-gyromagnetic-ratio hydrogen nucleus instead of the low-gyromagnetic-ratio nucleus that is bonded to the hydrogen atom.[105] In principle, hetereonuclear magnetization transfer MRI could be used to detect the presence or absence of specific chemical bonds.[106][107] NAWM and Diffusely abnormal areas (DAWM) appear under MT-MRI.
Finally, the fifth more important MRI technique is the Proton Magnetic resonance spectroscopy. Based in the different response to the electromagetic pulses that different substances present, a MRS scanner is able to identify chemical substances in the brain. This is important because N‐acetylaspartate is a marker of axonal damage that can be now identified in-vivo.[83]
Lesions under the special MRI methods
Normally two different kind of lesions appear on a normal MRI: T2-hypertense lesions and T1-hypointense. The first one are demyelinating lesions and appear brighter than the surroundings in T2-MRI.
The T1-hypointense are areas less dense than the surrounding NAW, and appear black on T1-MRI. They are mainly axonal degeneration areas. Because their black appearance they are sometimes known as black holes. They seem to appear as a sequel after a strong demyelinating lesion.
BBB disruption is normally shown using gadolinium. It is a contrast that cannot cross the BBB except when it is dysfunctional. Therefore, in active lesions with BBB implication the contrast enters the brain and appears in the MRI.
Before BBB disruption, some brain tissues which present normal aspect under T1 and T2 MRI (Normal appearing white matter, NAWM and normal appearing grey matter, NAGM), can show several abnormalities under special MRI technologies:
Magnetization transfer multi-echo T(2) relaxation. Subjects with Long-T(2) lesions had a significantly longer disease duration than subjects without this lesion subtype.[108] It has been found that grey matter injury correlates with disability[109] and that there is high oxidative stress in lesions, even in the old ones.[110]
Diffusion tensor MRI or Magnetic Transfer MRI are two options to enhance MRI-hidden abnormalities discovery. This is currently an active field of research with no definitive results, but it seems that these two technologies are complementary.[111]
Other methods of MRI allow us to get a better insight of the lesions structure. Recently MP-RAGE MRI has shown better results than PSIR and DIR for gray matter lesions.[112] Susceptibility weighted imaging (SWI-MRI) has shown iron (hemosiderin) deposition in lesions, and helps to detect otherwise invisible lesions.[113]
Abnormalities in the gray matter (Diffusion tensor MRI alterations) of the brain parenchyma are present early in the course of multiple sclerosis[114]
Normal appearing brain tissues
Using several texture analysis technologies, it is possible to classify white matter areas into three categories: normal, normal-appearing and lesions.[115] Currently, it is possible to detect lesions before they present demyelination, and they are called pre-active lesions.[101] A fourth area called DAWM (diffusely abnormal white matter) has recently been proposed[116] and can help to differentiate PPMS and SPMS.[117] Abundant extracellular myelin in the meninges of patients with multiple sclerosis has been found[118]
Brain tissues with MRI-hidden problems are usually named Normal Appearing. Exploring the normal-appearing corpus callosum has been found a possible primary hypoperfusion,[119][120] according with other findings in this same direction.[121][122][123][124][125][126] Also iron (in hemosiderin deposits and as well as in ferritin-like structures inside the macrophage) accumulation has been reported[127][128]
Several findings in these areas have been shown. Post-mortem studies over NAWM and NAGM areas (Normal appearing White and Gray Matters) show several biochemical alterations, like increased protein carbonylation and high levels of Glial fibrillary acidic protein (GFAP), which in NAGM areas comes together with higher than normal concentration of protein carbonyls, suggesting reduced levels of antioxidants and the presence of small lesions.[129] The amount of interneuronal Parvalbumin is lower than normal in brain's motor cortex areas,[130] and oxidative injury of oligodendrocytes and neurons could be associated with active demyelination and axonal injury.[131]
NAWM in MS has been reported to be similar to NAWM in leukoaraiosis,[132] though NAWM damage in MS is inflammatory and special microscopic techniques like CARS microscopy show that the CNS of MS patients may be globally altered, and both lesions and NAWM are just manifestations of another underlying problem.[133] The NAWM is specially abnormal close to the ventricles, which may indicate a pathogenic mechanism mediated via the CSF or ependyma.[134]
Non-lesional White Matter
Most of the brain in MS is unafected. Though obviously normal white matter appears normal under MRI, so does the NAWM white matter described in the next section. To establish a difference, normal white matter is named Non-lesional white matter (NLWM)[135]
This normal white matter is reported to be around 56% of the total WM of the patients.[136]
Normal appearing White Matter
The white matter with hidden but MRI-visible damage is known as "Normal-appearing white matter" (NAWM)[137] and is where lesions appear.[13] The NAWM is considered a non-visible kind of lesion, produces dissability and it is responsive to natalizumab[138]
The pathology of the NAWM differs from areas near the lesions or near the cortex. Close to WM lesions, axonal pathology and microglial activation may explain subtle MRI changes. Distant from lesions, microglial activation associated with proximity to cortical lesions might underlie MRI abnormalities.[139]
The NAWM precedes the lesions. It has been shown that the apparent diffusion coefficient (ADC) precedes the development of new plaques. Later increases during BBB breakdown (gadolinium enhancement) and finally decays after the enhancement.[140]
BBB disruption takes place on NAWM areas.[141] This can be read in different ways. Maybe some hidden changes in White Matter structure trigger the BBB disruption, or maybe the same process that creates the NAWM areas disrupts the BBB after some time.
Pre-active lesions are lesions in an early stage of development. They resolve sometimes without further damage, and not always develop into demyelinating lesions. They present clusters of activated microglia in otherwise normal-appearing white matter.[101][102]
Oligodendrocyte abnormalities appear to be crucially involved.[142][143] The earliest change reported in the lesions examined is widespread oligodendrocyte apoptosis in which T cells, macrophages, activated microglia, reactive astrocytes, and neurons appear normal. This observation points to some change in the local environment (NAWM) to which oligodendrocytes are especially susceptible and which triggers a form of apoptosis.[144]
Water diffusivity is higher in all NAWM regions, deep gray matter regions, and some cortical gray matter region of MS patients than normal controls.[145]
Citrullination appears in SPMS.[146] It seems that a defect of sphingolipid metabolism modifies the properties of normal appearing white matter.[147] Related to these, peptidylarginine deiminase 2 is increased in patients with MS, and is related to arginine de-imination.[148]
NAWM shows a decreased perfusion which does not appear to be secondary to axonal loss.[124] The reduced perfusion of the NAWM in MS might be caused by a widespread astrocyte dysfunction, possibly related to a deficiency in astrocytic beta(2)-adrenergic receptors and a reduced formation of cAMP, resulting in a reduced uptake of K(+) at the nodes of Ranvier and a reduced release of K(+) in the perivascular spaces.[149] This would be consistent again with cases of Chronic cerebrospinal venous insufficiency.
White matter lesions appear in NAWM areas,[13] and their behavior can be predicted by MRI parameters as MTR (magnetization transfer ratio).[150][151] This MTR parameter is related to axonal density.[152]
It also seems that myelin basic protein (MBP) from multiple sclerosis (MS) patients contains lower levels of phosphorylation at Thr97 than normal individuals.[153]
NAWM is the place where lesions appear and the process seems to be made by microglia, in absence of leukocyte infiltration, astrogliosis or demyelination. At the final stage of the process, these microglia develop into active demyelinating MS lesion[154]
Gray matter damage. Normal Appearing Gray Matter
Gray matter tissue damage dominates the pathological process as MS progresses, and underlies neurological disability. Imaging correlates of gray matter atrophy indicate that mechanisms differ in RRMS and SPMS.[155] Epstein-Barr virus could be involved,[156] but is not likely.[157] Involvement of the deep gray matter (DGM), suggested by magnetic resonance imaging, is confirmed, and most DGM lesions involve both GM and white matter. Inflammation in DGM lesions is intermediate between the destructive inflammation of white matter lesions and the minimal inflammation of cortical lesions.[158]
Iron depositions appear in deep gray matter by magnetic field correlation MRI[159] Differently from NAWM, NAGM areas are not related to the development of lesions[160]
Diffusely abnormal white matter
Other active area of study is the Diffusely abnormal white matter (DAWM). It seems to be a reduction of myelin phospholipids that correlates with a reduction of the myelin water fraction.[161] The DAWM consisted of extensive axonal loss, decreased myelin density, and chronic fibrillary gliosis, all of which were substantially abnormal compared with normal-appearing WM and significantly different from focal WM lesion pathology.[162] Changes in the vasculature take place not only in focal lesions but also in DAWM as detected by postmortem MRI[163]
Dirty appearing white matter
Dirty-appearing white matter (referred to as DAWM like the former case) is defined as a region with ill-defined borders of intermediate signal intensity between that of normal-appearing white matter (NAWM) and that of plaque on T2-weighted and proton density imaging.[164] It is probably created by loss of myelin phospholipids, detected by the short T2 component, and axonal reduction.
Microglial nodules
Originally proposed as a biomarker,[165] the presence of these nodules has a possible pathogenetic significance. Though their role in the lesion evolution is still unclear, their presence in normal-appearing white matter have been suggested to be an early stage of lesion formation [166]
Heterogeneity of the disease
Multiple sclerosis has been reported to be heterogeneus in its behavior, in its underlying mechanisms, in its response to medication [167] and remarkably, also respect the response to the specific potassium channel autoantibody Kir4.1.[168]
For some authors, what we call MS in reality is a heterogeneous group of diseases[169] Some independent reports take also PPMS apart[170]
Some reports propose the existence of molecular biomarkers that determine the clinical course of the disease,[171] but the relationship to the pathological types has still not been established as of 2016.
Demyelination patterns
Four different damage patterns have been identified in patients' brain tissues. The original report suggests that there may be several types of MS with different immune causes, and that MS may be a family of several diseases. Though originally was required a biopsy to classify the lesions of a patient, since 2012 it is possible to classify them by a blood test[172] looking for antibodies against seven lipids, three of which are cholesterol derivatives.[173]
It is believed that they may correlate with differences in disease type and prognosis, and perhaps with different responses to treatment. In any case, understanding lesion patterns can provide information about differences in disease between individuals and enable doctors to make more accurate treatment decisions
According to one of the researchers involved in the original research "Two patterns (I and II) showed close similarities to T-cell-mediated or T-cell plus antibody-mediated autoimmune encephalomyelitis, respectively. The other patterns (III and IV) were highly suggestive of a primary oligodendrocyte dystrophy, reminiscent of virus- or toxin-induced demyelination rather than autoimmunity."
The four identified patterns are:[174]
- Pattern I
- The scar presents T-cells and macrophages around blood vessels, with preservation of oligodendrocytes, but no signs of complement system activation.[175]
- Pattern II
- The scar presents T-cells and macrophages around blood vessels, with preservation of oligodendrocytes, as before, but also signs of complement system activation can be found.[176] This pattern has been considered similar to damage seen in NMO, though AQP4 damage does not appear in pattern II MS lesions[177] Nevertheless, pattern II has been reported to respond to plasmapheresis,[178] which points to something pathogenic into the blood serum.
- The complement system infiltration in these cases convert this pattern into a candidate for research into autoimmune connections like anti-Kir4.1,[179] anti-Anoctamin-2[180] or anti-MOG mediated MS[181] About the last possibility, research has found antiMOG antibodies in some pattern-II MS patients.[182]
- Sometimes autoimmunity against the human CNS has been triggered by accident or medical mistake. The reactions have been diverse according to the sources of the disease but pathological confirmed MS (damage fulfills all pathological criteria of MS) is among them, and it shows pattern II[183]
- Pattern II pathogenic T cells clonally expanded were found in the CN, specifically, CD4+ Th2 cells (secreting IL-4, L-5, and IL-13) have recently been described in pattern II MS, and their clones have been isolated as living cells[14][15][184] The functional characterization shows that T cells releasing Th2 cytokines and helping B cells dominate the T-cell infiltrate in pattern II brain lesions.[14]
- Pattern III
- The scars are diffuse with inflammation, distal oligodendrogliopathy and microglial activation. There is also loss of myelin-associated glycoprotein (MAG). The scars do not surround the blood vessels, and in fact, a rim of preserved myelin appears around the vessels. There is evidence of partial remyelinization and oligodendrocyte apoptosis. For some researchers this pattern is an early stage of the evolution of the others.[144] For others, it represents ischaemia-like injury with a remarkable availability of a specific biomarker in CSF[185][186]
- Some authors have stated that distal oligodendrogliopathy could come from a metabolic process[187]
- Pattern IV
- The scar presents sharp borders and oligodendrocyte degeneration, with a rim of normal appearing white matter. There is a lack of oligodendrocytes in the center of the scar. There is no complement activation or MAG loss.
These differences are noticeable only in early lesions[188] and the heterogeneity was controversial during some time because some research groups thought that these four patterns could be consequence of the age of the lesions.[189] Nevertheless, after some debate among research groups, the four patterns model is accepted and the exceptional case found by Prineas has been classified as NMO[190][191]
For some investigation teams this means that MS is a heterogeneous disease. Currently antibodies to lipids and peptides in sera, detected by microarrays, can be used as markers of the pathological subtype given by brain biopsy.[192]
Other developments in this area is the finding that some lesions present mitochondrial defects that could distinguish types of lesions.[193]
MRI Phenotypes
Several studies trying to stablish a relationship between the pathological findings and MRI findings have been performed.
For example, pulsed magnetization transfer imaging,[194] diffusion Tensor MRI,[195] and VCAM-1 enhanced MRI[196] have been reported to show the pathological differences of these patterns. Together with MRI, magnetic resonance spectroscopy allows to see the biochemical composition of the lesions, which shows at least two different patterns[197]
Currently as of 2014, the MRI studies have led to the proposal of four MRI phenotypes,[198] though both the classification and the relationship with the pathology remains controversial.
Other proposed correlations
Several correlations have been studied trying to stablish a pathological classification:
- With clinical courses: No definitive relationship between these patterns and the clinical subtypes has been established by now, but some relations have been established. All the cases with PPMS (primary progressive) had pattern IV (oligodendrocyte degeneration) in the original study[199] and nobody with RRMS was found with this pattern. Balo concentric sclerosis lesions have been classified as pattern III (distal oligodendrogliopathy).[200] Neuromyelitis optica was associated with pattern II (complement mediated demyelination), though they show a perivascular distribution, at difference from MS pattern II lesions.[201]
- With Optic Coherence Tomography: OCT of the retinal layer yields different results for PPMS and RRMS[202]
- With CSF findings: Teams in Oxford and Germany,[203] found correlation with CSF and progression in November 2001, and hypotheses have been made suggesting correlation between CSF findings and pathophysiological patterns.[204] In particular, B-cell to monocyte ratio looks promising.
The anti-MOG antibody has been investigated but no utility as biomarker has been found,[205] though this is disputed.[206] High levels of anti-nuclear antibodies are found normally in patients with MS. Antibodies against Neurofascins NF-155 [207] and NF-186 could be involved in subtypes of MS[208] Recently, it has been shown that the CSF from PPMS patients can transport the disease[170]
- With responses to therapy: It is known that 30% of MS patients are non-responsive to Beta interferon.[209] The heterogeneous response to therapy can support the idea of hetherogeneous aetiology. It has also been shown that IFN receptors and interleukins in blood serum predicts response to IFN therapy,[210][211] specially IL-17,[212] and interleukins IL12/IL10 ratio has been proposed as marker of clinical course.[213] Besides:
- Pattern II lesions patients are responsive to plasmapheresis, while others are not.[178][214]
- The subtype associated with macrophage activation, T cell infiltration and expression of inflammatory mediator molecules may be most likely responsive to immunomodulation with interferon-beta or glatiramer acetate.[215]
- People non-responsive to interferons are the most responsive to Copaxone [216]
- In general, people non-responsive to a treatment is more responsive to other,[217] and changing therapy can be effective.[218]
- There are genetic differences between responders and not responders.[219] Though the article points to heterogeneous metabolic reactions to interferons instead of disease heterogeneity, it has been shown that most genetic differences are not related to interferon behavior[220]
- With response to NMO-IgG:: NMO-IgG is the immunoglobulin that attacks Aquaporin-4 in Devic's disease. Multiple sclerosis patients do not have it in blood, but it has been shown that 13% of tested patients reacted with the epitope AQPaa252-275. It is not known if these antibodies define distinct MS subsets, or are simply markers of astrocytic damage
- With lesion structure: Cavitary lesions appear only in a subset of patients with a worse clinical course than normal[221]
- Response to intravenous immunoglobin: The response to IVIG is strongly dependent from the genetic profile of each person in a predictive way[222]
- Comorbidity with diabetes: Diabetes mellitus type 1 (T1D) is produced by special leukocyte antigen haplotypes, which seem to be involved also in some cases of MS[223]
Primary progressive MS
It is currently discussed whether Primary Progressive MS (PPMS) is a different pathological entity or a different degree of the same pathology. No agreement has been established but there are some pathological features that are specific to PPMS. For example, meningeal inflammation is different respect standard cases of Recurrent-Recidivant MS (RRMS)[224] and sodium accumulation is higher.[225] Diffusely Abnormal White Matter (DAWM) is different than in RRMS/SPMS patients[226] and it has been shown that CSF from PPMS patients can transport the disease[170]
From a pathological point of view, PPMS characteristics are slow expansion of pre-existing white matter lesions, massive cortical demyelination, and extensive diffuse injury of the normal appearing white matter. As in relapsing MS also in progressive MS active tissue injury is invariably associated with inflammation, but inflammation seems to be trapped behind a closed blood brain barrier[227]
Currently Natalizumab is being studied for PPMS[138]
Pathology of early MS and silent MS
McDonald criteria rely in detecting the lesions disseminated in time and space that define MS by clinical observations. Therefore, normally they do not allow to establish a diagnosis for definite MS before two clinical attacks have appeared. This means that for clinical definite cases, MS condition has been present for a long time, difficulting the study of the initial stages.[228] To study the initial stages of MS, some additional paraclinical tests must be used to prove the presence and dissemination of the lesions.[229]
Sometimes patients with their first isolated attack (Clinically Isolated syndrome, or CIS) but before the confirming second attack (Preclinical MS) can be accepted to study the initial MS pathology[230] but there is a study suggesting that any MS case begins as a silent pathology that can remain unnoticed even for five years.[231] Therefore, even the CIS can appear too late in MS evolution.
Cases of MS before the CIS are sometimes found during other neurological inspections and are referred to as subclinical MS.,[232] or sometimes Clinically silent MS.[233] The previous reference states that clinically silent MS plaques were located in the periventricular areas. This reference also reports an estimate of the prevalence of silent MS as high as about 25%. Oligodendrocytes evolution is similar to normal MS clinical courses[234]
Sometimes patients that undergo a MRI examination for an unrelated cause can show lesions in their brains. These cases of isolated MRI findings have been recently baptised as RIS (Radiologically Isolated Syndrome) and are the most common inspections in which suggestions of silent MS have appeared.[235]
In respect to the pathology of the RIS cases, we can point out that they show cortical lesions, mainly in patients with oligoclonal bands.[236] Macroscopic damage is similar to RRMS cases but milder.[237] Cervical cord lesions are an important predictor of progression[238] and the quotient N-acetylaspartate to creatine suggest axonal damage[239]
See also
References
- ↑ Lublin, F. D. (2016). "Multiple Sclerosis and Other Inflammatory Diseases". Mount Sinai Expert Guides: Neurology (eds S. C. Sealfon, R. Motiwala and C. B. Stacy) , John Wiley & Sons, Ltd, Chichester, UK.: 873–874. doi:10.1002/9781118621042.ch23.
- ↑ Dutta R1, Trapp BD. Pathology and definition of multiple sclerosis. Rev Prat. 2006 Jun 30;56(12):1293-8.
- ↑ Brosnan CF, Raine CS (2013). "The astrocyte in multiple sclerosis revisited". Glia. 61 (4): 453–465. doi:10.1002/glia.22443. PMID 23322421.
- ↑ Franciotta D, Salvetti M, Lolli F, Serafini B, Aloisi F. B cells and multiple sclerosis" Lancet Neurol 2008 Sep;7(9):852-8. doi:10.1016/S1474-4422(08)70192-3
- ↑ Meinl E, Krumbholz M, Derfuss T, Dewitt D,; Krumbholz; Derfuss; Junker; Hohlfeld (November 2008). "Compartmentalization of inflammation in the CNS: A major mechanism driving progressive multiple sclerosis". J Neurol Sci. 274 (1–2): 42–4. doi:10.1016/j.jns.2008.06.032. PMID 18715571.
- 1 2 Lassmann Hans (2014). "Multiple sclerosis: Lessons from molecular neuropathology". Experimental Neurology. 262: 2–7. doi:10.1016/j.expneurol.2013.12.003.
- ↑ Kutzelnigg Alexandra; et al. (2007). "Widespread Demyelination in the Cerebellar Cortex in Multiple Sclerosis". Brain Pathology. 17 (1): 38–44. doi:10.1111/j.1750-3639.2006.00041.x.
- ↑ Absinta M; et al. (Apr 2015). "Gadolinium-based MRI characterization of leptomeningeal inflammation in multiple sclerosis". Neurology. 85 (1): 18–28. doi:10.1212/WNL.0000000000001587.
- 1 2 Bogdan F. Gh. Popescu et al, Pathology of Multiple Sclerosis: Where Do We Stand? Continuum (Minneap Minn). 2013 Aug; 19(4 Multiple Sclerosis): 901–921. doi:10.1212/01.CON.0000433291.23091.65
- ↑ Haider Lukas; et al. "The topograpy of demyelination and neurodegeneration in the multiple sclerosis". Brain: awv398. doi:10.1093/brain/awv398.
- ↑ Nathan P. Young , Brian G. Weinshenker , Joseph E. Parisi , B. Scheithauer , C. Giannini , Shanu F. Roemer , Kristine M. Thomsen , Jayawant N. Mandrekar , Bradley J. Erickson , Claudia F. Lucchinetti, Perivenous demyelination: association with clinically defined acute disseminated encephalomyelitis and comparison with pathologically confirmed multiple sclerosis, Brain 2010, DOI:10.1093/brain/awp321 333-348 First published online: 3 February 2010
- ↑ Bogdan F. Popescu, MD, Istvan Pirko, and Claudia F. Lucchinetti, Pathology of Multiple Sclerosis: Where Do We Stand? Continuum (Minneap Minn). 2013 Aug; 19(4 Multiple Sclerosis): 901–921, PMCID:PMC3915566, doi:http://doi.org/10.1212/01.CON.0000433291.23091.65
- 1 2 3 Goodkin DE, Rooney WD, Sloan R, et al. (December 1998). "A serial study of new MS lesions and the white matter from which they arise". Neurology. 51 (6): 1689–97. doi:10.1212/wnl.51.6.1689. PMID 9855524.
- 1 2 3 4 Raquel Planas et al. Central role of Th2/Tc2 lymphocytes in pattern II multiple sclerosis lesions. 8 June 2015, doi:10.1002/acn3.218
- 1 2 Antel J. P.; Ludwin S. K.; Bar-Or A. (2015). "Sequencing the immunopathologic heterogeneity in multiple sclerosis". Annals of Clinical and Translational Neurology. 2: 873–874. doi:10.1002/acn3.230.
- ↑ Fransson ME, Liljenfeldt LS, Fagius J, Tötterman TH, Loskog AS (2009). "The T-cell pool is anergized in patients with multiple sclerosis in remission". Immunology. 126 (1): 92–101. doi:10.1111/j.1365-2567.2008.02881.x. PMC 2632699
 . PMID 18624727.
. PMID 18624727. - ↑ http://wwwchem.csustan.edu/chem4400/sjbr/corey02.htm
- ↑ Markovic-Plese S, Cortese I, Wandinger KP, McFarland HF, Martin R (October 2001). "CD4+CD28– costimulation-independent T cells in multiple sclerosis". J Clin Invest. 108 (8): 1185–94. doi:10.1172/JCI12516. PMC 209525. PMID 11602626.
- ↑ Wulff H, Calabresi PA, Allie R, et al. (June 2003). "The voltage-gated Kv1.3 K+ channel in effector memory T cells as new target for MS". J Clin Invest. 111 (11): 1703–13. doi:10.1172/JCI16921. PMC 156104. PMID 12782673.
- ↑ Rus H, et al. (August 2005). "The voltage-gated potassium channel Kv1.3 is highly expressed on inflammatory infiltrates in multiple sclerosis brain". Proc Natl Acad Sci USA. 102 (31): 11094–9. Bibcode:2005PNAS..10211094R. doi:10.1073/pnas.0501770102. PMC 1182417. PMID 16043714.
- ↑ Beeton C, Chandy KG (December 2005). "Potassium channels, memory T cells, and multiple sclerosis". Neuroscientist. 11 (6): 550–62. doi:10.1177/1073858405278016. PMID 16282596.
- ↑ Okuda Y, Okuda M, Apatoff BR, Posnett DN (August 2005). "The activation of memory CD4(+) T cells and CD8(+) T cells in patients with multiple sclerosis". J Neurol Sci. 235 (1–2): 11–7. doi:10.1016/j.jns.2005.02.013. PMID 15972217.
- ↑ Krakauer M, Sorensen PS, Sellebjerg F (December 2006). "CD4(+) memory T cells with high CD26 surface expression are enriched for Th1 markers and correlate with clinical severity of multiple sclerosis". J Neuroimmunol. 181 (1–2): 157–64. doi:10.1016/j.jneuroim.2006.09.006. PMID 17081623.
- ↑ Ratts RB, Karandikar NJ, Hussain RZ, et al. (September 2006). "Phenotypic characterization of autoreactive T cells in multiple sclerosis". J Neuroimmunol. 178 (1–2): 100–10. doi:10.1016/j.jneuroim.2006.06.010. PMID 16901549.
- ↑ Haegele KF, Stueckle CA, Malin JP, Sindern E (February 2007). "Increase of CD8+ T-effector memory cells in peripheral blood of patients with relapsing-remitting multiple sclerosis compared to healthy controls". J Neuroimmunol. 183 (1–2): 168–74. doi:10.1016/j.jneuroim.2006.09.008. PMID 17084910.
- ↑ Jilek S, Schluep M, Rossetti AO, et al. (April 2007). "CSF enrichment of highly differentiated CD8+ T cells in early multiple sclerosis". Clin Immunol. 123 (1): 105–13. doi:10.1016/j.clim.2006.11.004. PMID 17188575.
- ↑ Miyazaki Y, et al. (September 2008). "Expansion of CD4+CD28- T cells producing high levels of interferon-{gamma} in peripheral blood of patients with multiple sclerosis". Mult Scler. 14 (8): 1044–55. doi:10.1177/1352458508092809. PMID 18573819.
- ↑ Lünemann JD, Jelcić I, Roberts S, et al. (August 2008). "EBNA1-specific T cells from patients with multiple sclerosis cross react with myelin antigens and co-produce IFN-γ and IL-2". J Exp Med. 205 (8): 1763–73. doi:10.1084/jem.20072397. PMC 2525578. PMID 18663124.
- ↑ Hauser SL, Waubant E, Arnold DL, et al. (February 2008). "B-cell depletion with rituximab in relapsing-remitting multiple sclerosis". N Engl J Med. 358 (7): 676–88. doi:10.1056/NEJMoa0706383. PMID 18272891.
- 1 2 Cause of nerve fiber damage in multiple sclerosis identified
- ↑ Lisak RP, Benjamins JA, Nedelkoska L, Barger JL, Ragheb S, Fan B, Ouamara N, Johnson TA, Rajasekharan S, Bar-Or A (May 2012). "Secretory products of multiple sclerosis B cells are cytotoxic to oligodendroglia in vitro". J Neuroimmunol. 246 (1–2): 85–95. doi:10.1016/j.jneuroim.2012.02.015. PMID 22458983.
- 1 2 Pascual AM, Martínez-Bisbal MC, Boscá I, et al. (2007). "Axonal loss is progressive and partly dissociated from lesion load in early multiple sclerosis". Neurology. 69 (1): 63–7. doi:10.1212/01.wnl.0000265054.08610.12. PMID 17606882.
- ↑ Wolswijk G (15 January 1998). "Chronic stage multiple sclerosis lesions contain a relatively quiescent population of oligodendrocyte precursor cells". J Neurosci. 18 (2): 601–9. PMID 9425002.
- ↑ Jeroen J. G. Geurtsa, Lars Böc, Petra J. W. Pouwelsd, Jonas A. Castelijnsa, Chris H. Polmanb and Frederik Barkhof, Cortical Lesions in Multiple Sclerosis: Combined Postmortem MR Imaging and Histopathology, American Journal of Neuroradiology 26:572-577, March 2005
- ↑ Wattjes MP, Harzheim M, Kuhl CK, et al. (1 September 2006). "Does high-field MR imaging have an influence on the classification of patients with clinically isolated syndromes according to current diagnostic mr imaging criteria for multiple sclerosis?". Am J Neuroradiol. 27 (8): 1794–8. PMID 16971638.
- ↑ Nelson F, Poonawalla AH, Hou P, Huang F, Wolinsky JS, Narayana PA (October 2007). "Improved identification of intracortical lesions in multiple sclerosis with phase-sensitive inversion recovery in combination with fast double inversion recovery MR imaging". Am J Neuroradiol. 28 (9): 1645–9. doi:10.3174/ajnr.A0645. PMID 17885241.
- ↑ Roosendaal SD, Moraal B, Vrenken H, et al. (April 2008). "In vivo MR imaging of hippocampal lesions in multiple sclerosis". J Magn Reson Imaging. 27 (4): 726–31. doi:10.1002/jmri.21294. PMID 18302199.
- ↑ Geurts JJ, Pouwels PJ, Uitdehaag BM, Polman CH, Barkhof F, Castelijns JA (July 2005). "Intracortical lesions in multiple sclerosis: improved detection with 3D double inversion-recovery MR imaging". Radiology. 236 (1): 254–60. doi:10.1148/radiol.2361040450. PMID 15987979.
- ↑ Sampat MP, Berger AM, Healy BC, et al. (October 2009). "Regional White Matter Atrophy–Based Classification of Multiple Sclerosis in Cross-Sectional and Longitudinal Data". Am J Neuroradiol. 30 (9): 1731–9. doi:10.3174/ajnr.A1659. PMC 2821733. PMID 19696139.
- ↑ Gilmore CP, Donaldson I, Bö L, Owens T, Lowe JS, Evangelou N (October 2008). "Regional variations in the extent and pattern of grey matter demyelination in Multiple Sclerosis: a comparison between the cerebral cortex, cerebellar cortex, deep grey matter nuclei and the spinal cord". J Neurol Neurosurg Psychiatry. 80: 182–7. doi:10.1136/jnnp.2008.148767. PMID 18829630.
- ↑ Calabrese M, De Stefano N, Atzori M, et al. (2007). "Detection of cortical inflammatory lesions by double inversion recovery magnetic resonance imaging in patients with multiple sclerosis". Arch Neurol. 64 (10): 1416–22. doi:10.1001/archneur.64.10.1416. PMID 17923625.
- ↑ Poonawalla AH, Hasan KM, Gupta RK, et al. (2008). "Diffusion-Tensor MR Imaging of Cortical Lesions in Multiple Sclerosis: Initial Findings". Radiology. 246 (3): 880–6. doi:10.1148/radiol.2463070486. PMID 18195384.
- ↑ Calabrese M, Filippi M, Rovaris M, Mattisi I, Bernardi V, Atzori M, Favaretto A, Barachino L, Rinaldi L, Romualdi C, Perini P, Gallo P (2008). "Morphology and evolution of cortical lesions in multiple sclerosis. A longitudinal MRI study". NeuroImage. 42 (4): 1324–8. doi:10.1016/j.neuroimage.2008.06.028. PMID 18652903.
- ↑ Dawson fingers, at Radiopedia
- ↑ Agosta F, Pagani E, Caputo D, Filippi M (2007). "Associations between cervical cord gray matter damage and disability in patients with multiple sclerosis". Arch Neurol. 64 (9): 1302–5. doi:10.1001/archneur.64.9.1302. PMID 17846269.
- ↑ Agosta F, Valsasina P, Rocca MA, Caputo D, Sala S, Judica E, Stroman PW, Filippi M (2008). "Evidence for enhanced functional activity of cervical cord in relapsing multiple sclerosis". Magnetic Resonance in Medicine. 59 (5): 1035–42. doi:10.1002/mrm.21595. PMID 18429010.
- ↑ Cruz LC, Domingues RC, Gasparetto EL (June 2009). "Diffusion tensor imaging of the cervical spinal cord of patients with relapsing-remising multiple sclerosis: a study of 41 cases". Arq Neuropsiquiatr. 67 (2B): 391–5. doi:10.1590/S0004-282X2009000300004. PMID 19623432.
- ↑ Agosta F, Absinta M, Sormani MP, et al. (August 2007). "In vivo assessment of cervical cord damage in MS patients: a longitudinal diffusion tensor MRI study". Brain. 130 (Pt 8): 2211–9. doi:10.1093/brain/awm110. PMID 17535835.
- ↑ Gilmore C, Geurts J, Evangelou N, et al. (October 2008). "Spinal cord grey matter lesions in multiple sclerosis detected by post-mortem high field MR imaging". Multiple Sclerosis. 15 (2): 180–8. doi:10.1177/1352458508096876. PMID 18845658.
- ↑ Cornelia Laule et al. High-resolution myelin water imaging in post-mortem multiple sclerosis spinal cord: A case report, January 27, 2016, doi: 10.1177/1352458515624559, Mult Scler January 27, 2016 1352458515624559
- ↑ Redondo J1, Kemp K, Hares K, Rice C, Scolding N, Wilkins A. Purkinje cell pathology and loss in multiple sclerosis cerebellum. Brain Pathol. 2014 Nov 20. doi: 10.1111/bpa.12230. PMID 25411024
- ↑ Howell et al. Extensive grey matter pathology in the cerebellum in multiple sclerosis is linked to inflammation in the subarachnoid space, Neuropathol Appl Neurobiol. 2014 Nov; doi: 10.1111/nan.12199. PMID 25421634
- ↑ Romascano et al. Multicontrast connectometry: A new tool to assess cerebellum alterations in early relapsing-remitting multiple sclerosis, Hum Brain Mapp. 2014 Nov 24. doi: 10.1002/hbm.22698, PMID 25421928
- ↑ Deppe M; et al. (Apr 2015). "Evidence for early, non-lesional cerebellar damage in patients with multiple sclerosis: DTI measures correlate with disability, atrophy, and disease duration". Mult Scler. 22: 73–84. doi:10.1177/1352458515579439. PMID 25921041.
- ↑ Kipp M, Wagenknecht N, Beyer C, Samer S, Wuerfel J, Nikoubashman O (Nov 2014). "Thalamus pathology in multiple sclerosis: from biology to clinical application". Cell Mol Life Sci. 72: 1127–47. doi:10.1007/s00018-014-1787-9. PMID 25417212.
- 1 2 Kidd D.; Barkhof F.; McConnell R.; Algra P. R.; Allen I. V.; Revesz T. (1999). "Cortical lesions in multiple sclerosis". Brain. 122: 17–26. doi:10.1093/brain/122.1.17.
- ↑ Steenwijk MD; et al. (2015). "Unraveling the relationship between regional gray matter atrophy and pathology in connected white matter tracts in long-standing multiple sclerosis". Hum Brain Mapp. 36: 1796–1807. doi:10.1002/hbm.22738.
- ↑ Puthenparampil M; et al. (Mar 2015). "Cortical relapses in multiple sclerosis". Mult Scler. doi:10.1177/1352458514564483. PMID 25791367.
- ↑ Mistry Niraj; Abdel-Fahim Rasha; Gowland Penny (2014). "A CORTICOCENTRIC MODEL FOR MS PATHOGENESIS, J". Neurol Neurosurg Psychiatry. 85 (10): e4. doi:10.1136/jnnp-2014-309236.135.
- ↑ Mainero C; et al. (Feb 2015). "A gradient in cortical pathology in multiple sclerosis by in vivo quantitative 7 T imaging". Brain. 138: 932–45. doi:10.1093/brain/awv011. PMID 25681411.
- ↑ C. Mainero et al. Contribution of subpial pathology to cortical thinning in multiple sclerosis: a combined 7T - 3T MRI study, Proc. Intl. Soc. Mag. Reson. Med. 2010; 18
- ↑ Klaver R.; et al. (Apr 2015). "Neuronal and Axonal Loss in Normal-Appearing Gray Matter and Subpial Lesions in Multiple Sclerosis". J Neuropathol Exp Neurol. 74: 453–8. doi:10.1097/NEN.0000000000000189. PMID 25853695.
- ↑ Jürgens Tanja; Jafari Mehrnoosh; Kreutzfeldt Mario; Bahn Erik; Brück Wolfgang; Kerschensteiner Martin; Merkler Doron. "Reconstruction of single cortical projection neurons reveals primary spine loss in multiple sclerosis". Brain. 139: 39–46. doi:10.1093/brain/awv353.
- ↑ Richard Yates, Margaret Esiri, Jacqueline Palace, Gabriele De Luca. Fibrinogen Deposition in the Multiple Sclerosis Motor Cortex. Neurology April 5, 2016 vol. 86 no. 16 Supplement P1.369
- ↑ Gabriele De Luca et al. Right Under Our Noses: Olfactory Pathology In Central Nervous System Demyelinating Diseases, Neurology April 8, 2014 vol. 82 no. 10 Supplement P6.173
- ↑ Takahashi; et al. (Dec 2014). "Association of Cerebrospinal Fluid Levels of Lateral Olfactory Tract Usher Substance (LOTUS) With Disease Activity in Multiple Sclerosis". JAMA Neurol. 72: 176. doi:10.1001/jamaneurol.2014.3613.
- ↑ Frohman EM, Fujimoto JG, Frohman TC, Calabresi PA, Cutter G, Balcer LJ (December 2008). "Optical coherence tomography: a window into the mechanisms of multiple sclerosis". Nat Clin Pract Neurol. 4 (12): 664–75. doi:10.1038/ncpneuro0950. PMC 2743162. PMID 19043423.
- ↑ "eye, human."Encyclopædia Britannica. 2008. Encyclopædia Britannica 2006 Ultimate Reference Suite DVD
- ↑ Garcia-Martin E, Calvo B, Malvè M, Herrero R, Fuertes I, Ferreras A, Larrosa JM, Polo V, Pablo LE (2013). "Three-Dimensional Geometries Representing the Retinal Nerve Fiber Layer in Multiple Sclerosis, Optic Neuritis, and Healthy Eyes". Ophthal Res. 50: 72–81. doi:10.1159/000350413. PMID 23774269.
- ↑ Pulicken et al. Optical coherence tomography and disease subtype in multiple sclerosis. Neurology November 27, 2007 vol. 69 no. 22 2085-2092, DOI:10.1093/brain/awq080
- ↑ Pueyo V, Martin J, Fernandez J, Almarcegui C, Ara J, Egea C, Pablo L, Honrubia F (2008). "Axonal loss in the retinal nerve fiber layer in patients with multiple sclerosis". Multiple Sclerosis. 14 (5): 609–14. doi:10.1177/1352458507087326. PMID 18424482.
- ↑ Zaveri MS, Conger A, Salter A, Frohman TC, Galetta SL, Markowitz CE, Jacobs DA, Cutter GR, Ying GS, Maguire MG, Calabresi PA, Balcer LJ, Frohman EM (2008). "Retinal Imaging by Laser Polarimetry and Optical Coherence Tomography Evidence of Axonal Degeneration in Multiple Sclerosis". Archives of Neurology. 65 (7): 924–8. doi:10.1001/archneur.65.7.924. PMID 18625859.
- ↑ Sepulcre J, Murie-Fernandez M, Salinas-Alaman A, García-Layana A, Bejarano B, Villoslada P (May 2007). "Diagnostic accuracy of retinal abnormalities in predicting disease activity in MS". Neurology. 68 (18): 1488–94. doi:10.1212/01.wnl.0000260612.51849.ed. PMID 17470751.
- ↑ Naismith RT, Tutlam NT, Xu J, et al. (March 2009). "Optical coherence tomography differs in neuromyelitis optica compared with multiple sclerosis". Neurology. 72 (12): 1077–82. doi:10.1212/01.wnl.0000345042.53843.d5. PMC 2677471. PMID 19307541.
- ↑ Lucarelli MJ, Pepose JS, Arnold AC, Foos RY (November 1991). "Immunopathologic features of retinal lesions in multiple sclerosis". Ophthalmology. 98 (11): 1652–6. doi:10.1016/s0161-6420(91)32080-3. PMID 1724792.
- ↑ Kerrison JB, Flynn T, Green WR (1994). "Retinal pathologic changes in multiple sclerosis". Retina. 14 (5): 445–51. doi:10.1097/00006982-199414050-00010. PMID 7899721.
- ↑ Gugleta K, Kochkorov A, Kavroulaki D, et al. (April 2009). "Retinal vessels in patients with multiple sclerosis: baseline diameter and response to flicker light stimulation". Klin Monatsbl Augenheilkd. 226 (4): 272–5. doi:10.1055/s-0028-1109289. PMID 19384781.
- ↑ Kochkorov A, Gugleta K, Kavroulaki D, et al. (April 2009). "Rigidity of retinal vessels in patients with multiple sclerosis". Klin Monatsbl Augenheilkd. 226 (4): 276–9. doi:10.1055/s-0028-1109291. PMID 19384782.
- ↑ Ari J. Green, Stephen McQuaid, Stephen L. Hauser, Ingrid V. Allen, Roy Lyness, Ocular pathology in multiple sclerosis: retinal atrophy and inflammation irrespective of disease duration, DOI:10.1093/brain/awq080 1591-1601, 21 April 2010
- ↑ Gabilondo IM, Lapiscina EH, Heras E, Fraga PE, Llufriu S, Ortiz S, Villoslada P (2014). "Trans‐synaptic axonal degeneration in the visual pathway in multiple sclerosis". Annals of Neurology. 75 (1): 98–107. doi:10.1002/ana.24030.
- ↑ Maria; et al. (2013). "Wallerian and trans-synaptic degeneration contribute to optic radiation damage in multiple sclerosis: a diffusion tensor MRI study". Mult Scler. 19 (12): 1610–1617. doi:10.1177/1352458513485146.
- ↑ Optical Coherence Tomography in Multiple Sclerosis: Clinical Applications, Editor Axel Petzold, Springer, 28 ago. 2015 - 198 páginas
- 1 2 M. Filippi, M. Bozzali, M. Rovaris, O. Gonen, C. Kesavadas, A. Ghezzi, V. Martinelli, R. I. Grossman, G. Scotti, G. Comi, A. Falini. Evidence for widespread axonal damage at the earliest clinical stage of multiple sclerosis. DOI:10.1093/brain/awg038 433-437 First published online: 1 February 2003
- ↑ Huizinga R, Gerritsen W, Heijmans N, Amor S (September 2008). "Axonal loss and gray matter pathology as a direct result of autoimmunity to neurofilaments". Neurobiol Dis. 32 (3): 461–70. doi:10.1016/j.nbd.2008.08.009. PMID 18804534.
- ↑ Sobottka B, Harrer MD, Ziegler U, et al. (September 2009). "Collateral Bystander Damage by Myelin-Directed CD8+ T Cells Causes Axonal Loss". Am J Pathol. 175 (3): 1160–6. doi:10.2353/ajpath.2009.090340. PMC 2731134. PMID 19700745.
- ↑ Filippi M, Bozzali M, Rovaris M, Gonen O, Kesavadas C, Ghezzi A, Martinelli V, Grossman R, Scotti G, Comi G, Falini A (2003). "Evidence for widespread axonal damage at the earliest clinical stage of multiple sclerosis". Brain. 126 (Pt 2): 433–7. doi:10.1093/brain/awg038. PMID 12538409.
- ↑ Neuer Diagnose-Ansatz zur Früherkennung von MS
- ↑ Mostert JP, Blaauw Y, Koch MW, Kuiper AJ, Hoogduin JM, De Keyser J (2008). "Reproducibility over a 1-month period of 1H-MR spectroscopic imaging NAA/Cr ratios in clinically stable multiple sclerosis patients". Eur Radiol. 18 (8): 1736–40. doi:10.1007/s00330-008-0925-x. PMC 2469275. PMID 18389250.
- ↑ Franciotta D, Salvetti M, Lolli F, Serafini B, Aloisi F. B cells and multiple sclerosis. Lancet Neurol. 2008 Sep;7(9):852-8. doi: 10.1016/S1474-4422(08)70192-3.
- ↑ Cortical Lesions in Multiple Sclerosis, Inflammation versus Neurodegeneration, Hans Lassmann, Brain. 2012;135(10):2904-2905.
- ↑ Liu LP. Meningeal inflammation and multiple sclerosis. Neuroimmunol Neuroinflammation 2016;3:145-6. DOI:10.20517/2347-8659.2016.22
- ↑ Poser Charles M. "The peripheral nervous system in multiple sclerosis: A review and pathogenetic hypothesis". Journal of the Neurological Sciences. 79: 83–90. doi:10.1016/0022-510X(87)90262-0.
- ↑ Compston A, Coles A (October 2008). "Multiple sclerosis". Lancet. 372 (9648): 1502–17. doi:10.1016/S0140-6736(08)61620-7. PMID 18970977.
- ↑ Werring D. J. (2000). "The pathogenesis of lesions and normal-appearing white matter changes in multiple sclerosisA serial diffusion MRI study". Brain. 123 (8): 1667–76. doi:10.1093/brain/123.8.1667. PMID 10908196.
- ↑ Henderson AP, Barnett MH, Parratt JD, Prineas JW (December 2009). "Multiple sclerosis: distribution of inflammatory cells in newly forming lesions". Annals of Neurology. 66 (6): 739–53. doi:10.1002/ana.21800. PMID 20035511.
- ↑ Varun Sethi et al. Slowly eroding lesions in multiple sclerosis. Mult Scler June 23, 2016, doi:10.1177/1352458516655403
- ↑ van Walderveen MA; Kamphorst W; Scheltens P; van Waesberghe JH; Ravid R; Valk J; Polman CH; Barkhof F, Histopathologic correlate of hypointense lesions on T1-weighted spin-echo MRI in multiple sclerosis.
- ↑ Antonov SM, Kalinina NI, Kurchavyj GG, Magazanik LG, Shupliakov OV, Vesselkin NP (February 1990). "Identification of two types of excitatory monosynaptic inputs in frog spinal motoneurones". Neuroscience Letters. 109 (1–2): 82–7. doi:10.1016/0304-3940(90)90541-G. PMID 2156195.
- ↑ Guttmann Charles R. G.; Ahn Sungkee S.; Hsu Liangge; Kikinis Ron; Jolesz Ferenc A. (1995). "The Evolution of Multiple Sclerosis Lesions on Serial MR". Am J Neuroradiol. 16: 1481–1491.
- ↑ Gaitán María I; et al. (July 2011). "Evolution of the Blood-Brain Barrier in Newly Forming Multiple Sclerosis Lesions". Ann Neurol. 70 (1): 22–29. doi:10.1002/ana.22472.
- 1 2 3 van der Valk P, Amor S (June 2009). "Preactive lesions in multiple sclerosis". Current Opinion in Neurology. 22 (3): 207–13. doi:10.1097/WCO.0b013e32832b4c76. PMID 19417567.
- 1 2 Bsibsi M, Holtman IR, Gerritsen WH, Eggen BJ, Boddeke E, van der Valk P, van Noort JM, Amor S (Sep 2013). "Alpha-B-Crystallin Induces an Immune-Regulatory and Antiviral Microglial Response in Preactive Multiple Sclerosis Lesions". J Neuropathol Exp Neurol. 72: 970–9. doi:10.1097/NEN.0b013e3182a776bf. PMID 24042199.
- ↑ Laura E.; et al. "Jonkman et al. Can MS lesion stages be distinguished with MRI? A postmortem MRI and histopathology study". Journal of Neurology. 262 (4): 1074–1080. doi:10.1007/s00415-015-7689-4.
- ↑ Y. Ge, Multiple Sclerosis: The Role of MR Imaging, AJNR Am J Neuroradiol June 2006 27: 1165-1176
- ↑ Hurd RE, John BK (February 1991). "Gradient-enhanced proton-detected heteronuclear multiple-quantum coherence spectroscopy". Journal of Magnetic Resonance, Series A. 91 (3): 648–653. Bibcode:1991JMagR..91..648H. doi:10.1016/0022-2364(91)90395-a.
- ↑ Brown RA, Venters RA, Tang PP, Spicer LD (March 1995). "A test for scalar coupling between heteronuclei using gradient-enhanced proton-detected HMQC spectroscopy". Journal of Magnetic Resonance, Series A. 113 (1): 117–119. Bibcode:1995JMagR.113..117B. doi:10.1006/jmra.1995.1064.
- ↑ Miller AF, Egan LA, Townsend CA (March 1997). "Measurement of the degree of isotopic enrichment of different positions in an antibiotic peptide by NMR" (PDF). Journal of Magnetic Resonance. 125 (1): 120–131. Bibcode:1997JMagR.125..120M. doi:10.1006/jmre.1997.1107. PMID 9245367.
- ↑ Laule C, Vavasour IM, Kolind SH, et al. (2007). "Long T(2) water in multiple sclerosis: What else can we learn from multi-echo T(2) relaxation?". J Neurol. 254 (11): 1579–87. doi:10.1007/s00415-007-0595-7. PMID 17762945.
- ↑ Zhang Y, Zabad R, Wei X, Metz LM, Hill MD, Mitchell JR (2007). "Deep grey matter 'black T2' on 3 tesla magnetic resonance imaging correlates with disability in multiple sclerosis". Multiple Sclerosis. 13 (7): 880–3. doi:10.1177/1352458507076411. PMID 17468444.
- ↑ Holley JE, Newcombe J, Winyard PG, Gutowski NJ (2007). "Peroxiredoxin V in multiple sclerosis lesions: predominant expression by astrocytes". Multiple Sclerosis. 13 (8): 955–61. doi:10.1177/1352458507078064. PMID 17623739.
- ↑ Otaduy MC, Callegaro D, Bacheschi LA, Leite CC (December 2006). "Correlation of magnetization transfer and diffusion magnetic resonance imaging in multiple sclerosis". Multiple Sclerosis. 12 (6): 754–9. doi:10.1177/1352458506070824. PMID 17263003.
- ↑ Nelson F, Poonawalla A, Hou P, Wolinsky J, Narayana P (November 2008). "3D MPRAGE Improves Classification of Cortical Lesions in Multiple Sclerosis". Multiple Sclerosis. 14 (9): 1214–9. doi:10.1177/1352458508094644. PMC 2650249. PMID 18952832.
- ↑ Haacke EM, Makki M, Ge Y, et al. (March 2009). "Characterizing iron deposition in multiple sclerosis lesions using susceptibility weighted imaging". J Magn Reson Imaging. 29 (3): 537–44. doi:10.1002/jmri.21676. PMC 2650739. PMID 19243035.
- ↑ Cappellani R, Bergsland N, Weinstock-Guttman B3, Kennedy C2, Carl E, Ramasamy DP, Hagemeier J, Dwyer MG, Patti F, Zivadinov R "Diffusion tensor MRI alterations of subcortical deep gray matter in clinically isolated syndrome, J Neurol Sci. 2013 Dec; doi: 10.1016/j.jns.2013.12.031. PMID 24423584
- ↑ Zhang J, Tong L, Wang L, Li N (2008). "Texture analysis of multiple sclerosis: a comparative study". Magnetic Resonance Imaging. 26 (8): 1160–6. doi:10.1016/j.mri.2008.01.016. PMID 18513908.
- ↑ Seewann A, Vrenken H, van der Valk P, et al. (May 2009). "Diffusely abnormal white matter in chronic multiple sclerosis: imaging and histopathologic analysis". Arch Neurol. 66 (5): 601–9. doi:10.1001/archneurol.2009.57. PMID 19433660.
- ↑ Vrenken H, Seewann A, Knol DL, Polman CH, Barkhof F, Geurts JJ (March 2010). "Diffusely abnormal white matter in progressive multiple sclerosis: in vivo quantitative MR imaging characterization and comparison between disease types". American Journal of Neuroradiology. 31 (3): 541–8. doi:10.3174/ajnr.A1839. PMID 19850760.
- ↑ Kooi EJ, van Horssen J, Witte ME, et al. (June 2009). "Abundant extracellular myelin in the meninges of patients with multiple sclerosis". Neuropathol Appl Neurobiol. 35 (3): 283–95. doi:10.1111/j.1365-2990.2008.00986.x. PMID 19473295.
- ↑ A.M. Saindane, M. Law, Y. Ge, G. Johnson, J.S. Babb and R.I. Grossman 2007 Correlation of Diffusion Tensor and Dynamic Perfusion MR Imaging Metrics in Normal-Appearing Corpus Callosum: Support for Primary Hypoperfusion in Multiple Sclerosis, American Journal of Neuroradiology 28:767-772
- ↑ Juurlink BH (October 1998). "The multiple sclerosis lesion: initiated by a localized hypoperfusion in a central nervous system where mechanisms allowing leukocyte infiltration are readily upregulated?". Medical Hypotheses. 51 (4): 299–303. doi:10.1016/S0306-9877(98)90052-4. PMID 9824835.
- ↑ Inglese Matilde; Adhya Sumita; Johnson Glyn; Babb James S; Miles Laura; Jaggi Hina; Herbert Joseph; Grossman Robert. "Perfusion magnetic resonance imaging correlates of neuropsychological impairment in multiple sclerosis". Journal of Cerebral Blood Flow. 28: 164–171. doi:10.1038/sj.jcbfm.9600504.
- ↑ Adhya Sumita; Johnson Glyn; Herbert Joseph; Jaggi Hina; Babb James S.; Grossman Robert I.; Inglese Matilde. "Pattern of Hemodynamic Impairment in Multiple Sclerosis: Dynamic Susceptibility Contrast Perfusion MR Imaging at 3.0 T". NeuroImage. 33: 1029–1035. doi:10.1016/j.neuroimage.2006.08.008.
- ↑ Varga AW, Johnson G, Babb JS, Herbert J, Grossman RI, Inglese M (July 2009). "White Matter Hemodynamic Abnormalities precede Sub-cortical Gray Matter Changes in Multiple Sclerosis". J Neurol Sci. 282 (1–2): 28–33. doi:10.1016/j.jns.2008.12.036. PMC 2737614. PMID 19181347.
- 1 2 De Keyser J, Steen C, Mostert JP, Koch MW (October 2008). "Hypoperfusion of the cerebral white matter in multiple sclerosis: possible mechanisms and pathophysiological significance". Journal of Cerebral Blood Flow and Metabolism. 28 (10): 1645–51. doi:10.1038/jcbfm.2008.72. PMID 18594554.
- ↑ Inglese Matilde; Adhya Sumita; Johnson Glyn; Babb James S; Miles Laura; Jaggi Hina; Herbert Joseph; Grossman Robert I. "Perfusion magnetic resonance imaging correlates of neuropsychological impairment in multiple sclerosis". Journal of Cerebral Blood Flow. 28: 164–171. doi:10.1038/sj.jcbfm.9600504.
- ↑ Law M, Saindane AM, Ge Y, Babb JS, Johnson G, Mannon LJ, Herbert J, Grossman RI (June 2004). "Microvascular abnormality in relapsing-remitting multiple sclerosis: perfusion MR imaging findings in normal-appearing white matter". Radiology. 231 (3): 645–52. doi:10.1148/radiol.2313030996. PMID 15163806.
- ↑ Adams CW (February 1988). "Perivascular iron deposition and other vascular damage in multiple sclerosis". Journal of Neurology, Neurosurgery, and Psychiatry. 51 (2): 260–5. doi:10.1136/jnnp.51.2.260. PMC 1031540. PMID 3346691.
- ↑ Singh AV, Zamboni P (December 2009). "Anomalous venous blood flow and iron deposition in multiple sclerosis" (PDF). Journal of Cerebral Blood Flow and Metabolism. 29 (12): 1867–78. doi:10.1038/jcbfm.2009.180. PMID 19724286.
- ↑ Bizzozero OA, DeJesus G, Callahan K, Pastuszyn A (2005). "Elevated protein carbonylation in the brain white matter and gray matter of patients with multiple sclerosis". Journal of Neuroscience Research. 81 (5): 687–95. doi:10.1002/jnr.20587. PMID 16007681.
- ↑ Clements RJ, McDonough J, Freeman EJ (2008). "Distribution of parvalbumin and calretinin immunoreactive interneurons in motor cortex from multiple sclerosis post-mortem tissue". Experimental Brain Research. 187 (3): 459–65. doi:10.1007/s00221-008-1317-9. PMID 18297277.
- ↑ Haider Lukas; et al. "Oxidative damage in multiple sclerosis lesions". Brain. 134: 1914–1924. doi:10.1093/brain/awr128.
- ↑ Beggs Clive B (2013). "Venous hemodynamics in neurological disorders: an analytical review with hydrodynamic analysis". BMC Medicine. 11 (1): 142. doi:10.1186/1741-7015-11-142.

- ↑ Kelvin W. Poon et al. Quantitative biochemical investigation of various neuropathologies using high-resolution spectral CARS microscopy. Proc. SPIE 9305, Optical Techniques in Neurosurgery, Neurophotonics, and Optogenetics II, 930504 (March 10, 2015); doi:10.1117/12.2076654
- ↑ Chard Declan; et al. (2015). "Association of Multiple Sclerosis Normal Appearing White Matter Abnormality with Periventricular Location and Secondary Progression". Neurology. 84 (14 Suppl P6): 126.
- ↑ Petzold A, Tozer DJ, Schmierer K (Dec 2011). "Axonal damage in the making: neurofilament phosphorylation, proton mobility and magnetisation transfer in multiple sclerosis normal appearing white matter". Exp Neurol. 232 (2): 234–9. doi:10.1016/j.expneurol.2011.09.011.
- ↑ Barbosa S (1994). "Magnetic resonance relaxation time mapping in multiple sclerosis: Normal appearing white matter and the "invisible" lesion load". Magnetic Resonance Imaging. 12 (1): 33–42. doi:10.1016/0730-725x(94)92350-7.
- ↑ Mangia S, Carpenter AF, Tyan AE, Eberly LE, Garwood M, Michaeli S (Dec 2013). "Magnetization transfer and adiabatic T1ρ MRI reveal abnormalities in normal-appearing white matter of subjects with multiple sclerosis". Mult Scler. 20: 1066–1073. doi:10.1177/1352458513515084. PMID 24336350.
- 1 2 Wiebenga et al. White Matter Diffusion Changes during the First Year of Natalizumab Treatment in Relapsing-Remitting Multiple Sclerosis, AJNR, 2016
- ↑ Moll N. M.; Rietsch A. M.; Thomas S.; Ransohoff A. J.; Lee J.-C.; Fox R.; Chang A.; Ransohoff R. M.; Fisher E. (2011). "Multiple sclerosis normal-appearing white matter: Pathology–imaging correlations". Ann Neurol. 70 (5): 764–773. doi:10.1002/ana.22521.
- ↑ Werring DJ, Brassat D, Droogan AG, Clark CA, Symms MR, Barker GJ, MacManus DG, Thompson AJ, Miller DH., The pathogenesis of lesions and normal-appearing white matter changes in multiple sclerosis: a serial diffusion MRI study, NMR Research Unit, Queen Square, London, UK.
- ↑ Werring DJ, Brassat D, Droogan AG, et al. (August 2000). "The pathogenesis of lesions and normal-appearing white matter changes in multiple sclerosis: a serial diffusion MRI study". Brain. 123 (8): 1667–76. doi:10.1093/brain/123.8.1667. PMID 10908196.
- ↑ Allen; et al. (2001). "Pathological abnormalities in the normal-appearing white matter in multiple sclerosis". Neurol Sci. 22 (2): 141–4. doi:10.1007/s100720170012. PMID 11603615.
- ↑ Thomas Zeis; Ursula Graumann; Richard Reynolds; Nicole Schaeren-Wiemers (Jan 2008). "Normal-appearing white matter in multiple sclerosis is in a subtle balance between inflammation and neuroprotection". Brain. 131 (4): 288–303. doi:10.1093/brain/awm291. PMID 18056737.
- 1 2 Barnett MH, Prineas JW (April 2004). "Relapsing and remitting multiple sclerosis: pathology of the newly forming lesion" (PDF). Annals of Neurology. 55 (4): 458–68. doi:10.1002/ana.20016. PMID 15048884.
- ↑ Phuttharak W, Galassi W, Laopaiboon V, Laopaiboon M, Hesselink JR (2007). "Abnormal diffusivity of normal appearing brain tissue in multiple sclerosis: a diffusion-weighted MR imaging study". J Med Assoc Thai. 90 (12): 2689–94. PMID 18386722.
- ↑ Nicholas AP, Sambandam T, Echols JD, Tourtellotte WW (2004). "Increased citrullinated glial fibrillary acidic protein in secondary progressive multiple sclerosis". The Journal of Comparative Neurology. 473 (1): 128–36. doi:10.1002/cne.20102. PMID 15067723.
- ↑ Wheeler D, Bandaru VV, Calabresi PA, Nath A, Haughey NJ (November 2008). "A defect of sphingolipid metabolism modifies the properties of normal appearing white matter in multiple sclerosis". Brain. 131 (Pt 11): 3092–102. doi:10.1093/brain/awn190. PMC 2577809. PMID 18772223.
- ↑ Too Much Of A Charge-Switching Enzyme Causes Symptoms Of Multiple Sclerosis And Related Disorders In Mouse Models http://www.medicalnewstoday.com/articles/128393.php
- ↑ De Keyser J, Steen C, Mostert JP, Koch MW (2008). "Hypoperfusion of the cerebral white matter in multiple sclerosis: possible mechanisms and pathophysiological significance". Journal of Cerebral Blood Flow and Metabolism. 28 (10): 1645–51. doi:10.1038/jcbfm.2008.72. PMID 18594554.
- ↑ Filippi M, Rocca MA, Martino G, Horsfield MA, Comi G (June 1998). "Magnetization transfer changes in the normal appearing white matter precede the appearance of enhancing lesions in patients with multiple sclerosis". Annals of Neurology. 43 (6): 809–14. doi:10.1002/ana.410430616. PMID 9629851.
- ↑ Cercignani M, Iannucci G, Rocca MA, Comi G, Horsfield MA, Filippi M (March 2000). "Pathologic damage in MS assessed by diffusion-weighted and magnetization transfer MRI". Neurology. 54 (5): 1139–44. doi:10.1212/wnl.54.5.1139. PMID 10720288.
- ↑ van Waesberghe JH, Kamphorst W, De Groot CJ, et al. (November 1999). "Axonal loss in multiple sclerosis lesions: magnetic resonance imaging insights into substrates of disability". Annals of Neurology. 46 (5): 747–54. doi:10.1002/1531-8249(199911)46 (inactive 2015-02-01). PMID 10553992.
- ↑ Tait AR, Straus SK (August 2008). "Phosphorylation of U24 from Human Herpes Virus type 6 (HHV-6) and its potential role in mimicking myelin basic protein (MBP) in multiple sclerosis". FEBS Letters. 582 (18): 2685–8. doi:10.1016/j.febslet.2008.06.050. PMID 18616943.
- ↑ Shailender Singh, Imke Metz, Sandra Amor, Paul van der Valk, Christine Stadelmann, Wolfgang Brück, Microglial nodules in early multiple sclerosis white matter are associated with degenerating axons. Acta Neuropathologica, April 2013, Volume 125, Issue 4, pp 595-608
- ↑ Fisher E, Lee JC, Nakamura K, Rudick RA (September 2008). "Gray matter atrophy in multiple sclerosis: a longitudinal study". Annals of Neurology. 64 (3): 255–65. doi:10.1002/ana.21436. PMID 18661561.
- ↑ Zivadinov R, Zorzon M, Weinstock-Guttman B, et al. (June 2009). "Epstein-Barr virus is associated with grey matter atrophy in multiple sclerosis". J Neurol Neurosurg Psychiatr. 80 (6): 620–5. doi:10.1136/jnnp.2008.154906. PMID 19168469.
- ↑ Willis SN, Stadelmann C, Rodig SJ, et al. (July 2009). "Epstein–Barr virus infection is not a characteristic feature of multiple sclerosis brain". Brain. 132 (Pt 12): 3318–28. doi:10.1093/brain/awp200. PMC 2792367. PMID 19638446.
- ↑ Vercellino M, Masera S, Lorenzatti M, et al. (May 2009). "Demyelination, inflammation, and neurodegeneration in multiple sclerosis deep gray matter". J Neuropathol Exp Neurol. 68 (5): 489–502. doi:10.1097/NEN.0b013e3181a19a5a. PMID 19525897.
- ↑ Ge Y, Jensen JH, Lu H, et al. (October 2007). "Quantitative assessment of iron accumulation in the deep gray matter of multiple sclerosis by magnetic field correlation imaging". Am J Neuroradiol. 28 (9): 1639–44. doi:10.3174/ajnr.A0646. PMID 17893225.
- ↑ Capellani Roberto; et al. (2014). "Diffusion tensor MRI alterations of subcortical deep gray matter in clinically isolated syndrome". Journal of the Neurological Sciences. 338 (1–2): 128–134. doi:10.1016/j.jns.2013.12.031. PMID 24423584.
- ↑ Laule C, Vavasour IM, Leung E, Li DK, Kozlowski P, Traboulsee AL, Oger J, MacKay AL, Moore GW (October 2010). "Pathological basis of diffusely abnormal white matter: insights from magnetic resonance imaging and histology". Multiple Sclerosis. 17 (2): 144–50. doi:10.1177/1352458510384008. PMID 20965961.
- ↑ Seewann A, Vrenken H, van der Valk P, et al. (May 2009). "Diffusely abnormal white matter in chronic multiple sclerosis: imaging and histopathologic analysis". Arch Neurol. 66 (5): 601–9. doi:10.1001/archneurol.2009.57. PMID 19433660.
- ↑ Vos CM, Geurts JJ, Montagne L, et al. (December 2005). "Blood-brain barrier alterations in both focal and diffuse abnormalities on postmortem MRI in multiple sclerosis". Neurobiol Dis. 20 (3): 953–60. doi:10.1016/j.nbd.2005.06.012. PMID 16039866.
- ↑ Moore G. R. W.; Laule C.; MacKay A.; Leung E.; Li D. K. B.; Zhao G.; Traboulsee A. L.; Paty D. W. (2012). "Dirty-appearing white matter in multiple sclerosis". Journal of Neurology. 255 (11): 1802–1811. doi:10.1007/s00415-008-0002-z.
- ↑ Barnett MH, Parratt JD, Cho ES, Prineas JW (Jan 2009). "Immunoglobulins and complement in postmortem multiple sclerosis tissue". Ann Neurol. 65 (1): 32–46. doi:10.1002/ana.21524.
- ↑ Singh S, Metz I, Amor S, van der Valk P, Stadelmann C, Brück W (Apr 2013). "Microglial nodules in early multiple sclerosis white matter are associated with degenerating axons". Acta Neuropathol. 125 (4): 595–608. doi:10.1007/s00401-013-1082-0.
- ↑ Leussink VI, Lehmann HC, Meyer Zu Hörste G, Hartung HP, Stüve O, Kieseier BC (September 2008). "Rituximab induces clinical stabilization in a patient with fulminant multiple sclerosis not responding to natalizumab : Evidence for disease heterogeneity". J Neurology. 255 (9): 1436–8. doi:10.1007/s00415-008-0956-x. PMID 18685916.
- ↑ Rajneesh Srivastava et al. Potassium Channel KIR4.1 as an Immune Target in Multiple Sclerosis, New England Journal of Medicine, 2012; 367:115-123July 12, 2012DOI: 10.1056/NEJMoa1110740
- ↑ Alireza Minagar, Multiple Sclerosis: An Overview of Clinical Features, Pathophysiology, Neuroimaging, and Treatment Options (book), September 2014, 117 pages, (doi:10.4199/C00116ED1V01Y201408ISP055)
- 1 2 3 Cristofanilli Massimiliano; Rosenthal Hannah; Cymring Barbara; Gratch Daniel; Pagano Benjamin; Xie Boxun; Sadiq Saud A (2014). "Progressive multiple sclerosis cerebrospinal fluid induces inflammatory demyelination, axonal loss, and astrogliosis in mice". Experimental Neurology. 261: 620–632. doi:10.1016/j.expneurol.2014.07.020. PMID 25111532.
- ↑ Ahmed Enayetallah, Raghavendra Hosur, Richard Ransohoff and Jaya Goyal. Multiple Sclerosis Clinical Characteristics in Molecularly-Defined Patient Populations, Neurology April 5, 2016 vol. 86 no. 16 Supplement S37.008
- ↑ F. Quintana et al., Specific Serum Antibody Patterns Detected with Antigen Arrays Are Associated to the Development of MS in Pediatric Patients, Neurology, 2012. Freely available at
- ↑ Harnesing the clinical value of biomarkers in MS, International Journal of MS care, June 2012
- ↑ Lucchinetti CF, Brück W, Rodriguez M, Lassmann H (Jul 1996). "Distinct patterns of multiple sclerosis pathology indicates heterogeneity on pathogenesis". Brain Pathol. 6 (3): 259–74. doi:10.1111/j.1750-3639.1996.tb00854.x. PMID 8864283.
- ↑ Holmes, Nick (15 November 2001). "Part 1B Pathology: Lecture 11 - The Complement System". Retrieved 2006-05-10.
- ↑ Lucchinetti, Claudia; Wolfgang Brück; Joseph Parisi; Bernd Scheithauer; Moses Rodriguez; Hans Lassmann (December 1999). "A quantitative analysis of oligodendrocytes in multiple sclerosis lesions - A study of 113 cases". Brain. 122 (12): 2279–2295. doi:10.1093/brain/122.12.2279. PMID 10581222. Retrieved 2006-05-10.
- ↑ Kale N, Pittock SJ, Lennon VA, et al. (October 2009). "Humoral pattern II multiple sclerosis pathology not associated with neuromyelitis Optica IgG". Arch Neurol. 66 (10): 1298–9. doi:10.1001/archneurol.2009.199. PMC 2767176. PMID 19822791.
- 1 2 Wilner AN Goodman (March 2000). "Some MS patients have "Dramatic" responses to Plasma Exchange". Neurology Reviews. 8 (3).
- ↑ Srivastava R. et Al, Potassium channel KIR4.1 as an immune target in multiple sclerosis, N Engl J Med. 2012 Jul 12;367(2):115-23. doi: 10.1056/NEJMoa1110740, PMID 22784115
- ↑ Burcu Ayoglu et al. Anoctamin 2 identified as an autoimmune target in multiple sclerosis, February 9, 2016, doi: 10.1073/pnas.1518553113, PNAS February 23, 2016 vol. 113 no. 8 2188-2193
- ↑ Spadaro Melania; et al. (2015). "Histopathology and clinical course of MOG-antibody-associated encephalomyelitis". Annals of Clinical and Translational Neurology. 2 (3): 295–301. doi:10.1002/acn3.164.
- ↑ Jarius S, Metz I, König FB, Ruprecht K, Reindl M, Paul F, Brück W, Wildemann B.Screening for MOG-IgG and 27 other anti-glial and anti-neuronal autoantibodies in 'pattern II multiple sclerosis' and brain biopsy findings in a MOG-IgG-positive case. Mult Scler. 2016 Feb 11. pii: 1352458515622986.
- ↑ Höftberger R1, Leisser M2, Bauer J2, Lassmann H3. Autoimmune encephalitis in humans: how closely does it reflect multiple sclerosis? Acta Neuropathol Commun. 2015 Dec 4;3(1):80. doi: 10.1186/s40478-015-0260-9. PMID 26637427
- ↑ Martin R, Sospedra M, Rosito M, Engelhardt B (2016). "Current multiple sclerosis treatments have improved our understanding of MS autoimmune pathogenesis". Eur. J. Immunol. 46: 2078–2090. doi:10.1002/eji.201646485.
- ↑ Hans Lassmann et al. A new paraclinical CSF marker for hypoxia‐like tissue damage in multiple sclerosis lesions, Oxford Journals Medicine Brain Volume 126, Issue 6 Pp. 1347-1357
- ↑ Christina Marik , Paul A. Felts , Jan Bauer , Hans Lassmann , Kenneth J. Smith, Lesion genesis in a subset of patients with multiple sclerosis: a role for innate immunity? Brain, DOI:10.1093/brain/awm236 2800-2815
- ↑ Qiao Ling Cui et al. Oligodendrogliopathy in Multiple Sclerosis: Relation to Low Glycolytic Metabolic Rate of Oligodendrocytes (I10.004), Neurology April 5, 2016 vol. 86 no. 16 Supplement I10.004
- ↑ Breij EC, Brink BP, Veerhuis R, et al. (2008). "Homogeneity of active demyelinating lesions in established multiple sclerosis". Annals of Neurology. 63 (1): 16–25. doi:10.1002/ana.21311. PMID 18232012.
- ↑ Michael H. Barnett; John W. Prineas (2004). "Relapsing and Remitting Multiple Sclerosis: Pathology of the Newly Forming Lesion" (PDF). Annals of Neurology. 55 (1): 458–468. doi:10.1002/ana.20016. PMID 15048884.
- ↑ Brück W, Popescu B, Lucchinetti CF, Markovic-Plese S, Gold R, Thal DR, Metz I (Sep 2012). "Neuromyelitis optica lesions may inform multiple sclerosis heterogeneity debate". Ann Neurol. 72 (3): 385–94. doi:10.1002/ana.23621.
- ↑ Arnold P, Mojumder D, Detoledo J, Lucius R, Wilms H (Feb 2014). "Pathophysiological processes in multiple sclerosis: focus on nuclear factor erythroid-2-related factor 2 and emerging pathways". Clin Pharmacol. 6: 35–42. doi:10.2147/CPAA.S35033. PMID 24591852.
- ↑ Quintana FJ, et al. (December 2008). "Antigen microarrays identify unique serum autoantibody signatures in clinical and pathologic subtypes of multiple sclerosis". Proc Natl Acad Sci USA. 105 (48): 18889–94. Bibcode:2008PNAS..10518889Q. doi:10.1073/pnas.0806310105. PMC 2596207. PMID 19028871.
- ↑ Mahad D, Ziabreva I, Lassmann H, Turnbull D (2008). "Mitochondrial defects in acute multiple sclerosis lesions". Brain. 131 (Pt 7): 1722–35. doi:10.1093/brain/awn105. PMC 2442422. PMID 18515320.
- ↑ Smith SA, Farrell JA, Jones CK, Reich DS, Calabresi PA, van Zijl PC (October 2006). "Pulsed magnetization transfer imaging with body coil transmission at 3 Tesla: feasibility and application". Magn Reson Med. 56 (4): 866–75. doi:10.1002/mrm.21035. PMID 16964602.
- ↑ Goldberg-Zimring D, Mewes AU, Maddah M, Warfield SK (2005). "Diffusion tensor magnetic resonance imaging in multiple sclerosis". J Neuroimaging. 15 (4 Suppl): 68S–81S. doi:10.1177/1051228405283363. PMID 16385020.
- ↑ New imaging technique allows doctors to ‘see’ molecular activity
- ↑ West J, Aalto A, Tisell A, Leinhard OD, Landtblom AM, Smedby O, Lundberg P (2014). "Normal Appearing and Diffusely Abnormal White Matter in Patients with Multiple Sclerosis Assessed with Quantitative MR.". PLOS ONE. 9: e95161. doi:10.1371/journal.pone.0095161. PMC 3991609. PMID 24747946.
- ↑ Tauhid Shahamat; Neema Mohit; Healy Brian C.; Weiner Howard L.; Bakshi Rohit. "MRI phenotypes based on cerebral lesions and atrophy in patients with multiple sclerosis". Journal of Neurological Sciences. 346: 250–254. doi:10.1016/j.jns.2014.08.047.
- ↑ Primary progressive multiple sclerosis
- ↑ (Article in Spanish) Estudio longitudinal mediante imagen de resonancia magnética (RM) del efecto de la azatioprina
- ↑ The Mystery of the Multiple Sclerosis Lesion, Frontiers Beyond the Decade of the Brain, Medscape
- ↑ Balk L, Tewarie P, Killestein J, Polman C, Uitdehaag B, Petzold A. Disease course heterogeneity and OCT in multiple sclerosis. Mult Scler. 2014 Jan 8
- ↑ Cepok S, Jacobsen M, Schock S, et al. (November 2001). "Patterns of cerebrospinal fluid pathology correlate with disease progression in multiple sclerosis". Brain. 124 (Pt 11): 2169–76. doi:10.1093/brain/124.11.2169. PMID 11673319.
- ↑ Cepok S, Jacobsen M, Schock S, et al. (November 2001). "Patterns of cerebrospinal fluid pathology correlate with disease progression in multiple sclerosis". Brain. 124 (Pt 11): 2169–76. doi:10.1093/brain/124.11.2169. PMID 11673319.
- ↑ Pittock SJ, Reindl M, Achenbach S, et al. (January 2007). "Myelin oligodendrocyte glycoprotein antibodies in pathologically proven multiple sclerosis: frequency, stability and clinicopathologic correlations". Multiple Sclerosis. 13 (1): 7–16. doi:10.1177/1352458506072189. PMID 17294606.
- ↑ Belogurov AA, Kurkova IN, Friboulet A, et al. (January 2008). "Recognition and degradation of myelin basic protein peptides by serum autoantibodies: novel biomarker for multiple sclerosis". Journal of Immunology. 180 (2): 1258–67. doi:10.4049/jimmunol.180.2.1258. PMID 18178866.
- ↑ O. Stich; S. Perera; B. Berger; S. Jarius; B. Wildemann; A. Baumgartner; S. Rauer (March 2016). "Prevalence of neurofascin-155 antibodies in patients with multiple sclerosis". Journal of the neurological sciences. 364: 29–32. doi:10.1016/j.jns.2016.03.004.
- ↑ Early research into a treatment for progressive MS
- ↑ Fernández O, Fernández V, Mayorga C, et al. (December 2005). "HLA class II and response to interferon-beta in multiple sclerosis". Acta Neurol Scand. 112 (6): 391–4. doi:10.1111/j.1600-0404.2005.00415.x. PMID 16281922.
- ↑ van Baarsen LG, et al. (2008). Lassmann H, ed. "Pharmacogenomics of Interferon-β Therapy in Multiple Sclerosis: Baseline IFN Signature Determines Pharmacological Differences between Patients". PLOS ONE. 3 (4): e1927. Bibcode:2008PLoSO...3.1927V. doi:10.1371/journal.pone.0001927. PMC 2271130. PMID 18382694.
- ↑ Wiesemann E, Deb M, Hemmer B, Radeke HH, Windhagen A (2008). "Early identification of interferon-beta responders by ex vivo testing in patients with multiple sclerosis". Clinical Immunology. 128 (3): 306–13. doi:10.1016/j.clim.2008.04.007. PMID 18539537.
- ↑ Axtell RC; et al. (April 2010). "T helper type 1 and 17 cells determine efficacy of interferon-beta in multiple sclerosis and experimental encephalomyelitis". Nat Med. 16: 406–12. doi:10.1038/nm.2110. PMC 3042885. PMID 20348925.
- ↑ Carrieri PB, Ladogana P, Di Spigna G, et al. (2008). "Interleukin-10 and interleukin-12 modulation in patients with relapsing-remitting multiple sclerosis on therapy with interferon-beta 1a: differences in responders and non responders". Immunopharmacol Immunotoxicol. 30 (4): 1–9. doi:10.1080/08923970802302753. PMID 18686100.
- ↑ Patients' Multiple Sclerosis Lesion Type Dictates Effective Treatment
- ↑ Bitsch A, Brück W (2002). "Differentiation of multiple sclerosis subtypes: implications for treatment". CNS Drugs. 16 (6): 405–18. doi:10.2165/00023210-200216060-00004. PMID 12027786.
- ↑ Debouverie M, Moreau T, Lebrun C, Heinzlef O, Brudon F, Msihid J (November 2007). "A longitudinal observational study of a cohort of patients with relapsing-remitting multiple sclerosis treated with glatiramer acetate". Eur J Neurol. 14 (11): 1266–74. doi:10.1111/j.1468-1331.2007.01964.x. PMID 17956447.
- ↑ Carrá A, Onaha P, Luetic G, et al. (2008). "Therapeutic outcome 3 years after switching of immunomodulatory therapies in patients with relapsing-remitting multiple sclerosis in Argentina". Eur J Neurol. 15 (4): 386–93. doi:10.1111/j.1468-1331.2008.02071.x. PMID 18353125.
- ↑ Gajofatto A, Bacchetti P, Grimes B, High A, Waubant E (October 2008). "Switching first-line disease-modifying therapy after failure: impact on the course of relapsing-remitting multiple sclerosis". Multiple Sclerosis. 15 (1): 50–8. doi:10.1177/1352458508096687. PMID 18922831.
- ↑ Byun E, Caillier SJ, Montalban X, et al. (March 2008). "Genome-wide pharmacogenomic analysis of the response to interferon beta therapy in multiple sclerosis". Arch Neurol. 65 (3): 337–44. doi:10.1001/archneurol.2008.47. PMID 18195134.
- ↑ Vandenbroeck K, Matute C (May 2008). "Pharmacogenomics of the response to IFN-beta in multiple sclerosis: ramifications from the first genome-wide screen". Pharmacogenomics. 9 (5): 639–45. doi:10.2217/14622416.9.5.639. PMID 18466107.
- ↑ Corlobé A et al. Cavitary lesions in multiple sclerosis: Multicenter study on twenty patients, Rev Neurol (Paris). 2013 Oct 17. pii: S0035-3787(13)00939-9. doi: 10.1016/j.neurol.2013.02.010, PMID 24139243
- ↑ Berger; et al. (Oct 2014). "Predicting therapeutic efficacy of intravenous immunoglobulin (IVIG) in individual patients with relapsing remitting multiple sclerosis (RRMS) by functional genomics". J Neuroimmunol. 277 (1-2): 145–152. doi:10.1016/j.jneuroim.2014.10.001. PMID 25454729.
- ↑ Tettey et al. The co-occurrence of multiple sclerosis and type 1 diabetes: Shared aetiologic features and clinical implication for MS aetiology. J Neurol Sci. 2014 Nov; doi: 10.1016/j.jns.2014.11.019 PMID 25480016
- ↑ Choi SR, Howell OW, Carassiti D, Magliozzi R, Gveric D, Muraro PA, Nicholas R, Roncaroli F, Reynolds R (October 2012). "Meningeal inflammation plays a role in the pathology of primary progressive multiple sclerosis". Brain. 135: 2925–37. doi:10.1093/brain/aws189. PMID 22907116.
- ↑ Paling D, Solanky BS, Riemer F, Tozer DJ, Wheeler-Kingshott CA, Kapoor R, Golay X, Miller DH (Jul 2013). "Sodium accumulation is associated with disability and a progressive course in multiple sclerosis". Brain. 136: 2305–17. doi:10.1093/brain/awt149. PMID 23801742.
- ↑ Vrenken H.; et al. (2010). ", Diffusely Abnormal White Matter in Progressive Multiple Sclerosis: In Vivo Quantitative MR Imaging Characterization and Comparison between Disease Types". AJNR. 31: 541–548. doi:10.3174/ajnr.A1839. PMID 19850760.
- ↑ Lassmann H (Nov 2009). "Clinical and pathological topics of multiple sclerosis". Rinsho Shinkeigaku. 49 (11): 715–8. doi:10.5692/clinicalneurol.49.715. PMID 20030193.
- ↑ Frisullo G, Nociti V, Iorio R, et al. (December 2008). "The persistency of high levels of pSTAT3 expression in circulating CD4+ T cells from CIS patients favors the early conversion to clinically defined multiple sclerosis". J Neuroimmunol. 205 (1–2): 126–34. doi:10.1016/j.jneuroim.2008.09.003. PMID 18926576.
- ↑ Lassmann H (2010). "Acute disseminated encephalomyelitis and multiple sclerosis". Brain. 133 (2): 317–319. doi:10.1093/brain/awp342. PMID 20129937.
- ↑ Lebrun C, Bensa C, Debouverie M, et al. (2008). "Unexpected multiple sclerosis: follow-up of 30 patients with magnetic resonance imaging and clinical conversion profile". J Neurol Neurosurg Psychiatry. 79 (2): 195–198. doi:10.1136/jnnp.2006.108274. PMID 18202208.
- ↑ Nakamura M, Morris M, Cerghet M, Schultz L, Elias S (Fall 2014). "Longitudinal Follow-up of a Cohort of Patients with Incidental Abnormal Magnetic Resonance Imaging Findings at Presentation and Their Risk of Developing Multiple Sclerosis". Int J MS Care. 16 (3): 111–5. doi:10.7224/1537-2073.2013-016. PMC 4204370. PMID 25337052.
- ↑ Hakiki B, Goretti B, Portaccio E, Zipoli V, Amato MP (2008). "Subclinical MS: follow-up of four cases". European Journal of Neurology. 15 (8): 858–61. doi:10.1111/j.1468-1331.2008.02155.x. PMID 18507677.
- ↑ Engell T (May 1989). "A clinical patho-anatomical study of clinically silent multiple sclerosis". Acta Neurol Scand. 79 (5): 428–30. doi:10.1111/j.1600-0404.1989.tb03811.x. PMID 2741673.
- ↑ Mews I, Bergmann M, Bunkowski S, Gullotta F, Brück W (April 1998). "Oligodendrocyte and axon pathology in clinically silent multiple sclerosis lesions". Mult Scler. 4 (2): 55–62. doi:10.1177/135245859800400203. PMID 9599334.
- ↑ Siva A (Dec 2013). "Asymptomatic MS.". Clin Neurol Neurosurg. 115 (Suppl 1): S1–5. doi:10.1016/j.clineuro.2013.09.012. PMID 24321147.
- ↑ Giorgio A; et al. (Nov 2011). "Cortical lesions in radiologically isolated syndrome". Neurology. 77 (21): 1896–9. doi:10.1212/WNL.0b013e318238ee9b. PMID 22076541.
- ↑ De Stefano N; et al. (Apr 2011). "Improving the characterization of radiologically isolated syndrome suggestive of multiple sclerosis". PLOS ONE. 6 (4): e19452. doi:10.1371/journal.pone.0019452.
- ↑ Granberg T; et al. (Mar 2013). "Radiologically isolated syndrome--incidental magnetic resonance imaging findings suggestive of multiple sclerosis, a systematic review". Mult Scler. 19 (3): 271–80. doi:10.1177/1352458512451943.
- ↑ Stromillo ML; et al. (Jun 2013). "Brain metabolic changes suggestive of axonal damage in radiologically isolated syndrome". Neurology. 80 (23): 2090–4. doi:10.1212/WNL.0b013e318295d707.
External links
- The lesion project page
- MRI and CT of Multiple Sclerosis MedPix Image Database