Aspartoacylase
| Aspartoacylase | |||||||||
|---|---|---|---|---|---|---|---|---|---|
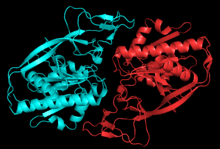 Structure of aspartoacylase dimer. Generated from 2I3C.[1] | |||||||||
| Identifiers | |||||||||
| EC number | 3.5.1.15 | ||||||||
| Databases | |||||||||
| IntEnz | IntEnz view | ||||||||
| BRENDA | BRENDA entry | ||||||||
| ExPASy | NiceZyme view | ||||||||
| KEGG | KEGG entry | ||||||||
| MetaCyc | metabolic pathway | ||||||||
| PRIAM | profile | ||||||||
| PDB structures | RCSB PDB PDBe PDBsum | ||||||||
| |||||||||
Aspartoacylase (EC 3.5.1.15, aminoacylase II, N-acetylaspartate amidohydrolase, acetyl-aspartic deaminase, acylase II, ASPA) is a hydrolase enzyme responsible for catalyzing the deacylation of N-acetyl-l-aspartate (N-acetylaspartate,NAA) into aspartate and acetate.[2][3] It is a zinc-dependent hydrolase that promotes the deprotonation of water to use as a nucleophile in a mechanism analogous to many other zinc-dependent hydrolases.[4] It is most commonly found in the brain, where it controls the levels of N-actetyl-l-aspartate. Mutations that result in loss of aspartoacylase activity are associated with Canavan disease, a rare autosomal recessive neurodegenerative disorder.[5]
Structure
Aspartoacylase is a dimer of two identical monomers of 313 amino acids and use a zinc cofactor in each.[1][6] There are two distinct domains in each monomer: the N-terminal domain from residues 1-212 and the C-terminal domain from residues 213-313.[7] The N-terminal domain of aspartoacylase is similar to that of zinc-dependent hydrolases such as carboxypeptidaseA. However, carboxypeptidases do not have something similar to the C-domain. In carboxypeptidase A, the active site is accessible to large substrates like the bulky C-terminal residue of polypeptides, whereas the C-domain sterically hinders access to the active site in aspartoacylase. Instead, the N-domain and C-domain of aspartoacylase form a deep narrow channel that leads to the active site.[1]
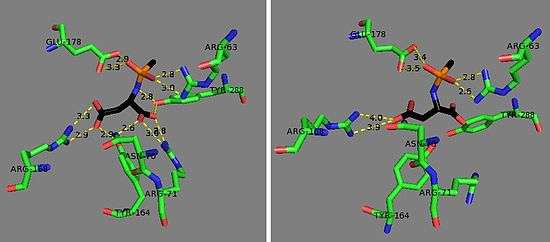
The zinc cofactor is found at the active site and is held by Glu-24, His-21, and His 116.[8] The substrate is held in place by Arg-63, Asn-70, Arg-71, Tyr-164, Arg-168, and Tyr-288.[1] The zinc cofactor is used to lower the pKa of a ligated water so that an attack on N-acetyl-L-aspartate may occur and to stabilize the resulting tetrahedral intermediate along with Arg-63, and Glu-178.[8]

Mechanism
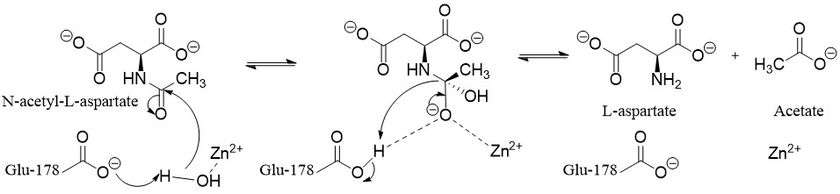
There are two types of possible mechanisms for zinc-dependent hydrolases depending on what is the nucleophile. The first uses deprotonated water and the second attacks with an asparate or glutamate first forming an anhydride.[9] Aspartoacylase follows the deprotonated water mechanism.[8] Zinc lowers the pKa of a ligated water molecule and the reaction proceeds via an attack on N-acetyl-l-aspartate when the water molecule is deprotonated by Glu-178.[1] This leads to a tetrahedral intermediate that is stabilized by the zinc, Arg-63, and Glu-178.[8] Finally, the carbonyl is then reformed, the bond with nitrogen is broken, and the nitrogen is protonated by the proton taken by Glu-178 all in one concerted step.[9]
Biological Function
Aspartoacylase is used to metabolize N-acetyl-L-aspartate by catalyzing its deacylation. Aspartoacylase prevents the build up of N-acetyl-L-aspartate in the brain. N-acetyl-L-aspartate is one of the most abundant amino acids found in the brain with concentrations of up to 10mM and makes up about 1% of the brain's dry weight.[10] It is believed that controlling N-acetyl-L-aspartate levels is essential for developing and maintaining white matter.[1] It is not known why so much N-acetyl-L-aspartate is produced in the brain nor what its primary function is.[11] However, one hypothesis is that it is potentially used as a chemical reservoir that can be tapped into for acetate for acetyl-CoA synthesis or aspartate for glutamate synthesis.[10][11][12] This way, N-acetyl-L-aspartate can be used to transport these precursor molecules and aspartoacylase is used to release them. For example, N-acetyl-L-aspartate produced in neurons can be transported into oligodendrocytes and the acetate released can be used for myelin synthesis.[7][13] Another hypothesis is that N-acetyl-L-aspartate is essential osmolyte that acts as a molecular water pump that helps maintain a proper fluid balance in the brain.[14]
Disease Relevance
Mutations that lead to loss of aspartoacylase activity have been identified as the cause of Canavan disease.[15] Canavan disease is a rare autosomal recessive disorder that causes spongy degeneration of the white matter in the brain and severe psychomotor retardation, usually leading to death at a young age.[7][16] The loss of aspartoacylase activity leads to the buildup of N-acetyl-L-aspartate in the brain and an increase in urine concentration by up to 60 times normal levels.[15] Though the exact mechanism of how loss of aspartoacylase activity leads to Canavan disease is not fully understood, there are two primary competing explanations.The first is that it leads to defective myelin synthesis due to a deficiency of acetyl-CoA derived from the acetate product.[16] Another explanation is that the elevated levels of N-acetyl-l-aspartate interfere with its normal brain osmoregulatory mechanism leading to osmotic disequilibrium.[17]
There are over 70 reported mutations of this enzyme, but the most common ones are the amino acid substitutions E285A and A305E.[7] E285A reduces activity of aspartoacylase down to as low as 0.3% of its normal function and is found in 98% of cases with Ashkenazi Jewish ancestry.[18] The mutation A305E is found in about 40% of non-Jewish patients and reduces activity to about 10%.[18] Of these two mutations, a crystal structure of the E285A mutant has been taken, showing that the loss of the hydrogen bonding from glutamate leads to a conformational change that distorts the active site and alters the substrate binding, leading to the much lower catalytic activity.[7]
References
- 1 2 3 4 5 6 7 8 Bitto, E; Bingman, CA; Wesenberg, GE; McCoy, JG; Phillips GN, Jr (9 January 2007). "Structure of aspartoacylase, the brain enzyme impaired in Canavan disease.". Proceedings of the National Academy of Sciences of the United States of America. 104 (2): 456–61. doi:10.1073/pnas.0607817104. PMC 1766406
 . PMID 17194761.
. PMID 17194761. - ↑ Birnbaum, S.M. (1955). "Aminoacylase. Amino acid aminoacylases I and II from hog kidney". Methods Enzymol. 2: 115–119. doi:10.1016/S0076-6879(55)02176-9.
- ↑ Birnbaum, S.M.; Levintow, L.; Kingsley, R.B.; Greenstein, J.P. (1952). "Specificity of amino acid acylases". J. Biol. Chem. 194: 455–470. PMID 14927637.
- ↑ Le Coq, J; An, HJ; Lebrilla, C; Viola, RE; Snee, K; Gorton, R; Smart, S (9 May 2006). "Characterization of human aspartoacylase: the brain enzyme responsible for Canavan disease.". Biochemistry. 45 (18): 5878–84. doi:10.1021/bi052608w. PMC 2566822. PMID 16669630.
- ↑ Hershfield, JR; Pattabiraman, N; Madhavarao, CN; Namboodiri, MA (7 May 2007). "Mutational analysis of aspartoacylase: implications for Canavan disease.". Brain Research. 1148: 1–14. doi:10.1016/j.brainres.2007.02.069. PMC 1933483. PMID 17391648.
- ↑ Herga, S; Berrin, JG; Perrier, J; Puigserver, A; Giardina, T (30 October 2006). "Identification of the zinc binding ligands and the catalytic residue in human aspartoacylase, an enzyme involved in Canavan disease.". FEBS Letters. 580 (25): 5899–904. doi:10.1016/j.febslet.2006.09.056. PMID 17027983.
- 1 2 3 4 5 6 Wijayasinghe, YS; Pavlovsky, AG; Viola, RE (5 August 2014). "Aspartoacylase catalytic deficiency as the cause of Canavan disease: a structural perspective.". Biochemistry. 53 (30): 4970–8. doi:10.1021/bi500719k. PMID 25003821.
- 1 2 3 4 5 6 Le Coq, J; Pavlovsky, A; Malik, R; Sanishvili, R; Xu, C; Viola, RE (18 March 2008). "Examination of the mechanism of human brain aspartoacylase through the binding of an intermediate analogue.". Biochemistry. 47 (11): 3484–92. doi:10.1021/bi702400x. PMC 2666850. PMID 18293939.
- 1 2 Zhang, Chenghua; Liu, Xiaoqiang; Xue, Ying (January 2012). "A general acid–general base reaction mechanism for human brain aspartoacylase: A QM/MM study". Computational and Theoretical Chemistry. 980: 85–91. doi:10.1016/j.comptc.2011.11.023.
- 1 2 Clark, JF; Doepke, A; Filosa, JA; Wardle, RL; Lu, A; Meeker, TJ; Pyne-Geithman, GJ (2006). "N-acetylaspartate as a reservoir for glutamate.". Medical Hypotheses. 67 (3): 506–12. doi:10.1016/j.mehy.2006.02.047. PMID 16730130.
- 1 2 Moffett, JR; Arun, P; Ariyannur, PS; Namboodiri, AM (26 December 2013). "N-Acetylaspartate reductions in brain injury: impact on post-injury neuroenergetics, lipid synthesis, and protein acetylation.". Frontiers in neuroenergetics. 5: 11. doi:10.3389/fnene.2013.00011. PMC 3872778. PMID 24421768.
- ↑ Hershfield, JR; Madhavarao, CN; Moffett, JR; Benjamins, JA; Garbern, JY; Namboodiri, A (October 2006). "Aspartoacylase is a regulated nuclear-cytoplasmic enzyme.". FASEB Journal. 20 (12): 2139–41. doi:10.1096/fj.05-5358fje. PMID 16935940.
- ↑ Chakraborty, G; Mekala, P; Yahya, D; Wu, G; Ledeen, RW (August 2001). "Intraneuronal N-acetylaspartate supplies acetyl groups for myelin lipid synthesis: evidence for myelin-associated aspartoacylase.". Journal of Neurochemistry. 78 (4): 736–45. doi:10.1046/j.1471-4159.2001.00456.x. PMID 11520894.
- ↑ Baslow, MH (April 2002). "Evidence supporting a role for N-acetyl-L-aspartate as a molecular water pump in myelinated neurons in the central nervous system. An analytical review.". Neurochemistry international. 40 (4): 295–300. doi:10.1016/s0197-0186(01)00095-x. PMID 11792458.
- 1 2 Moore, RA; Le Coq, J; Faehnle, CR; Viola, RE (1 May 2003). "Purification and preliminary characterization of brain aspartoacylase.". Archives of Biochemistry and Biophysics. 413 (1): 1–8. doi:10.1016/s0003-9861(03)00055-9. PMID 12706335.
- 1 2 Namboodiri, AM; Peethambaran, A; Mathew, R; Sambhu, PA; Hershfield, J; Moffett, JR; Madhavarao, CN (27 June 2006). "Canavan disease and the role of N-acetylaspartate in myelin synthesis.". Molecular and cellular endocrinology. 252 (1-2): 216–23. doi:10.1016/j.mce.2006.03.016. PMID 16647192.
- ↑ Baslow, MH; Guilfoyle, DN (April 2013). "Canavan disease, a rare early-onset human spongiform leukodystrophy: insights into its genesis and possible clinical interventions.". Biochimie. 95 (4): 946–56. doi:10.1016/j.biochi.2012.10.023. PMID 23151389.
- 1 2 Zano, S; Wijayasinghe, YS; Malik, R; Smith, J; Viola, RE (January 2013). "Relationship between enzyme properties and disease progression in Canavan disease.". Journal of Inherited Metabolic Disease. 36 (1): 1–6. doi:10.1007/s10545-012-9520-z. PMID 22850825.
External links
- aspartoacylase at the US National Library of Medicine Medical Subject Headings (MeSH)