Acetoacetate decarboxylase
| Acetoacetate decarboxylase | |||||||||
|---|---|---|---|---|---|---|---|---|---|
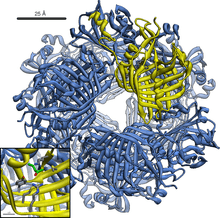 Acetoacetate decarboxylase dodecamer structure with bound 2-Pentanone bound in its active sites. | |||||||||
| Identifiers | |||||||||
| EC number | 4.1.1.4 | ||||||||
| CAS number | 9025-03-0 | ||||||||
| Databases | |||||||||
| IntEnz | IntEnz view | ||||||||
| BRENDA | BRENDA entry | ||||||||
| ExPASy | NiceZyme view | ||||||||
| KEGG | KEGG entry | ||||||||
| MetaCyc | metabolic pathway | ||||||||
| PRIAM | profile | ||||||||
| PDB structures | RCSB PDB PDBe PDBsum | ||||||||
| Gene Ontology | AmiGO / EGO | ||||||||
| |||||||||
Acetoacetate decarboxylase (AAD or ADC) is an enzyme involved in both the ketone body production pathway in humans and other mammals, and solventogenesis in bacteria. Acetoacetate decarboxylase plays a key role in solvent production by catalyzing the decarboxylation of acetoacetate, yielding acetone and carbon dioxide.[1] This enzyme has been of particular interest because it is a classic example of how pKa values of ionizable groups in the enzyme active site can be significantly perturbed. Specifically, the pKa value of lysine 115 in the active site is unusually low, allowing for the formation of a Schiff base intermediate and catalysis.[2]
| acetoacetic acid | Acetoacetate decarboxylase | acetone | |
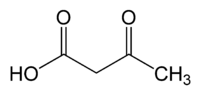 |
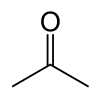 | ||
| CO2 | |||
| | |||
History
Acetoacetate decarboxylase is an enzyme with major historical implications, specifically in World War I and in establishing the state of Israel.[3] During the war the Allies needed pure acetone as a solvent for nitro-cellulose, a highly flammable compound that is the main component in gunpowder.[4] In 1916, biochemist and future first president of Israel Chaim Weizmann was the first to isolate Clostridium acetobutylicum, a Gram-positive, anaerobic bacteria in which acetoacetate decarboxylase is found. Weizmann was able to harness the organism’s ability to yield acetone from starch in order to mass-produce explosives during the war.[3] This led the American and British governments to install the process devised by Chaim Weizmann in several large plants in England, France, Canada, and the United States. Through Weizmann’s scientific contributions in World War I, he became close with influential British leaders educating them of his Zionist beliefs.[5] One of them was Arthur Balfour, the man after whom the Balfour Declaration—the first document pronouncing British support in the establishment of a Jewish homeland—was named.
The production of acetone by acetoacetate decarboxylase-containing or clostridial bacteria was utilized in large-scale industrial syntheses in the first half of the twentieth century. In the 1960s, the industry replaced this process with less expensive, more efficient chemical syntheses of acetone from petroleum and petroleum derivatives.[6] However, there has been a growing interest in acetone production that is more environmentally friendly, causing a resurgence in utilizing acetoacetate decarboxylase-containing bacteria.[7] Similarly, isopropanol and butanol fermentation using clostridial species is also becoming popular.
Structure
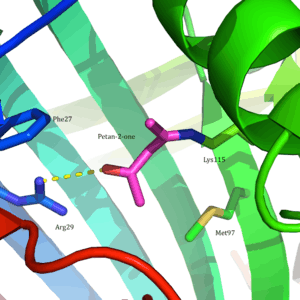
Acetoacetate decarboxylase is a 365 kDa complex with a homododecameric structure.[8] The overall structure consists of antiparallel β-sheets and a central seven-stranded cone-shaped β-barrel. The core of this β-barrel surrounds the active site in each protomer of the enzyme. The active site, consisting of residues such as Phe27, Met97, and Tyr113, is mostly hydrophobic. However, the active site does contain two charged residues: Arg29 and Glu76. Arg29 is thought to play a role in substrate binding, while Glu76 is thought to play a role in the orienting the active site for catalysis. The overall hydrophobic environment of the active site plays a critical role in favoring the neutral amine form of Lys115, a key residue involved in the formation of a Schiff base intermediate. Another important lysine residue, Lys116, is thought to play an important role in the positioning of Lys115 in the active site. Through hydrogen bonds with Ser16 and Met210, Lys116 positions Lys115 in the hydrophobic pocket of the active site to favor the neutral amine form.
Reaction mechanism
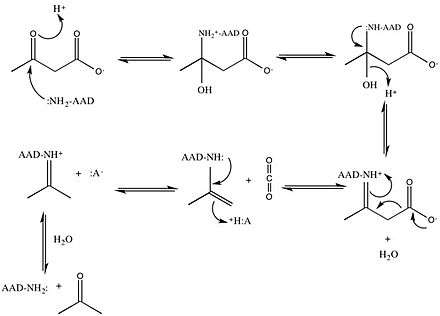
Acetoacetate decarboxylase from Clostridium acetobutylicum catalyzes the decarboxylation of acetoacetate to yield acetone and carbon dioxide (Figure 1). The reaction mechanism proceeds via the formation of a Schiff base intermediate, which is covalently attached to lysine 115 in the active site. The first line of support for this mechanism came from a radiolabeling experiment in which researchers labeled the carbonyl group of acetoacetate with 18O and observed that oxygen exchange to water, used as the solvent, is a necessary part of decarboxylation step.[9] These results provided support that the mechanism proceeds through a Schiff base intermediate between the ketoacid and an amino acid residue on the enzyme.
Further research led to the isolation of an active site peptide sequence and identification of the active site lysine, Lys115, that is involved in the formation of the Schiff base intermediate.[2][10] Additionally, later experiments led to the finding that maximum activity of the enzyme occurs at pH 5.95, suggesting that the pKa of the ε-ammonium group of Lys115 is significantly perturbed in the active site.[2] If the pKa were not perturbed downward, the lysine residue would remain protonated as an ammonium cation, making it unreactive for the nucleophilic addition necessary to form the Schiff base.
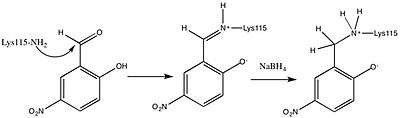
Building upon this finding, Westheimer et al. directly measured the pKa of Lys115 in the active site using 5-nitrosalicylaldehyde (5-NSA) . Reaction of 5-NSA with acetoacetate decarboxylase and subsequent reduction of the resulting Schiff base with sodium borohydride led to the incorporation of a 2-hydroxy-5-nitrobenzylamino reporter molecule in the active site (Figure 2). Titration of the enzyme with this attached reporter group revealed that the pKa of Lys115 is decreased to 5.9 in the active site.[12] These results were the basis for the proposal that the perturbation in the pKa of Lys115 was due to its proximity to the positively charged ε-ammonium group of Lys116 in the active site.[2] A nearby positive charge could cause unfavorable electrostatic repulsions that weaken the N-H bond of Lys115. Westheimer et al.'s proposal was further supported by site-directed mutagenesis studies. When Lys116 was mutated to cysteine or asparagine, the pKa of Lys115 was found to be significantly elevated to over 9.2, indicating that positively charged Lys116 plays a critical role in determining the pKa of Lys115.[2] Although a crystal structure was not yet solved to provide structural evidence, this proposal was widely accepted and cited as a textbook example of how the active site can be precisely organized to perturb a pKa and affect reactivity.[8]
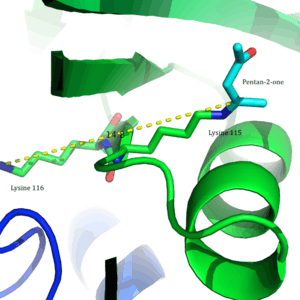
In 2009, a crystal structure of acetoacetate decarboxylase from Clostridium acetobutylicum was solved, allowing Westheimer et al.'s proposal to be evaluated from a new perspective . From the crystal structure, researchers found that Lys 115 and Lys 116 are oriented in opposite directions and separated by 14.8 Å (Figure 3).[8] This distance makes it unlikely that the positive charge of Lys116 is able to affect the pKa of Lys115. Instead through hydrogen bonds with Ser16 and Met210, Lys116 likely holds Lys115 into position in a hydrophobic pocket of the active site. This positioning disrupts the stability of the protonated ammonium cation of Lys115, suggesting that the perturbation of Lys115's pKa occurs through a 'desolvation effect'.
Inactivation and inhibition
Acetoacetate decarboxylase is inhibited by a number of compounds. Acetic anhydride performs an electrophilic attack on the critical catalytic residue, Lys115, of acetoacetate decarboxylase to inactivate the enzyme.[13] The rate of inactivation was assessed through the hydrolysis of the synthetic substrate 2,4-dinitrophenyl propionate to dinitrophenol by acetoacetate decarboxylase. In the presence of acetic anhydride, the enzyme is inactivated, unable to catalyze the hydrolysis reaction 2,4-dinitrophenyl propionate to dinitrophenol.[14]
Acetonylsulfonate acts as a competitive inhibitor (KI=8.0 mM ) as it mimics the characteristics of the natural substrate, acetoacetate (KM=8.0 mM).[15] The monoanion version of acetonylphosphonate is also a good inhibitor (KI=0.8mM), more efficient than the acetonylphosphonate monoester or dianion.[16] These findings indicate that active site is very discriminatory and sterically restricted.
Hydrogen cyanide seems to be an uncompetitive inhibitor, combining with Schiff’s base compounds formed at the active site.[15] Addition of carbonyl compounds to the enzyme, in the presence of hydrogen cyanide, increases hydrogen cyanide’s ability to inhibit acetoacetate decarboxylase, suggesting that carbonyl compounds readily form Schiff’s bases at the active site. Hydrogen cyanide is most potent as an inhibitor at pH 6, the optimum pH for the enzyme, suggesting that the rate-limiting step of catalysis is the formation of the Schiff base intermediate.
Beta-diketones appear to inhibit acetoacetate decarboxylase well but slowly. Acetoacetate decarboxylase has a KM for acetoacetate of 7×10−3 M whereas the enzyme has a KI for benzoylacetone of 1.9×10−6 M.[15] An enamine is most likely formed upon interaction of beta-diketones with free enzyme.
The reaction of acetoacetate decarboxylase with p-chloromercuriphenylsulfonate (CMS) results in decreased catalytic activity upon two equivalents of CMS per enzyme subunit.[15] CMS interacts with two sulfhydryl groups located on each enzyme subunit. Further inactivation occurs upon addition of a third equivalent of CMS per subunit. Addition of free cysteine to the inhibited enzyme is able to reverse CMS inhibition of acetoacetate decarboxylase.
Activity in bacteria
Acetoacetate decarboxylase has been found and studied in the following bacteria in addition to Clostridium acetobutylicum:
- Bacillus polymyxa
- Chromobacterium violaceum
- Clostridium beijerinckii
- Clostridium cellulolyticum
- Pseudomonas putida
Activity in humans and mammals
While this enzyme has not been purified from human tissue, the activity was shown to be present in human blood serum.[17][18]
In humans and other mammals, the conversion of acetoacetate into acetone and carbon dioxide by acetoacetate decarboxylase is a final irreversible step in the ketone-body pathway that supplies the body with a secondary source of energy.[19] In the liver, acetyl co-A formed from fats and lipids are transformed into three ketone bodies: acetone, acetoacetate, and D-β-hydroxybutyrate. Acetoacetate and D-β-hydroxybutyrate are exported to non-hepatic tissues, where they are converted back into acetyl-coA and used for fuel. Acetone and carbon dioxide on the other hand are exhaled, and not allowed to accumulate under normal conditions.
Acetoacetate and D-β-hydroxybutyrate freely interconvert through the action of D-β-hydroxybutyrate dehydrogenase.[19] Subsequently, one function of acetoacetate decarboxylase may be to regulate the concentrations of the other, two 4-carbon ketone bodies.
Clinical significance
Ketone body production increases significantly when the rate of glucose metabolism is insufficient in meeting the body's energy needs. Such conditions include high-fat ketogenic diets, diabetic ketoacidosis, or severe starvation.[20]
Under elevated levels of acetoacetate and D-β-hydroxybutyrate, acetoacetate decarboxylase produces significantly more acetone. Acetone is toxic, and can accumulate in the body under these conditions. Elevated levels of acetone in the human breath can be used to diagnose diabetes.[20]
References
- ↑ Peterson DJ, Bennett GN (1990). "Purification of acetoacetate decarboxylase from Clostridium acetobutylicum ATCC cloning of the acetoacetate decarboxylase in Escherichia coli.". Applied and Environmental Microbiology. 56 (11): 3491–3498.
- 1 2 3 4 5 6 Highbarger, LA; JA Gerlt; GL Kenyon (January 9, 1996). "Mechanism of the reaction catalyzed by acetoacetate decarboxylase. Importance of lysine 116 in determining the pKa of active site lysine 115". Biochemistry. 9 (35): 41–46. doi:10.1021/bi9518306. PMID 8555196.
- 1 2 Bormon, S (2009). "New Structure Revisits History". Structural Biology. 87 (21): 9.
- ↑ "Britannica Online".
- ↑ "Jewish Virtual Library".
- ↑ "Modeling of ABE Fermentation" (PDF).
- ↑ Collas, Florent; Wouter Kuit; Benjamin Clement; Remy Marchal; Ana M Lopez-Contreras; Frederic Monot (August 21, 2012). "Simultaneous production of isoproponal, butanol, ethanol, and 2,3-butanediol by Clostridium acetobutylicum ATCC 824 engineered strains". AMB Express. 2 (1): 45. doi:10.1186/2191-0855-2-45. PMC 3583297
 . PMID 22909015.
. PMID 22909015. - 1 2 3 4 5 Ho MC, Ménétret JF, Tsuruta H, Allen KN (May 21, 2009). "The origin of the electrostatic perturbation in acetoacetate decarboxylase". Nature. 459: 393–397. doi:10.1038/nature07938. PMID 19458715.
- ↑ Hamilton GA, Westheimer FH (1959). "On the Mechanism of Enzymatic Decarboxylation of Acetoacetate". J. Am. Chem. Soc. 81 (23): 6332–6333. doi:10.1021/ja01532a058.
- ↑ Warren, Stuart; Burt Zerner, F.H. Westheimer (March 1966). "Acetoacetate Decarboxylase. Identification of Lysine at the Active Site". Biochemistry. 5 (3): 817–823. doi:10.1021/bi00867a002.
- ↑ "A History of Acetoacetate Decarboxylase". JinKai.org. Retrieved 26 May 2014.
- ↑ Kokesh, Fritz C.; F.H. Westheimer (December 29, 1971). "A reporter group at the active site of acetoacetate decarboxylase. Ionization constant of the amino group.". Journal of the American Chemical Society. 93 (26): 7270–7274. doi:10.1021/ja00755a025.
- ↑ O'Leary, M.H.; F.H. Westheimer (1968). "Acetoacetate decarboxylase. Selective acetylation of enzyme". Biochemistry. 7 (3): 913–919. doi:10.1021/bi00843a005.
- ↑ Schmidt, Donald E.; F.H. Westheimer (1971). "pK of the Lysine Amino Group at the Active Site of Acetoacetate Decarboxylase". Biochemistry. 10 (7): 1249–1253. doi:10.1021/bi00783a023.
- 1 2 3 4 Autor, Anne P.; I. Fridovich (1970). "The Interactions of Acetoacetate Decarboxylase with Carbonyl Compounds, Hydrogen Cyanide, and an Organic Mercurial" (PDF). J. Biol. Chem. 245 (20): 5214–5222.
- ↑ Kluger, Ronald; Kurt Nakaoka (1974). "Inhibition of acetoacetate decarboxylase by ketophosphonates. Structural and dynamic probes of the active site". Biochemistry. 13 (5): 910–914. doi:10.1021/bi00702a013.
- ↑ van Stekelenburg GJ, Koorevaar G (June 1972). "Evidence for the existence of mammalian acetoacetate decarboxylase: with special reference to human blood serum". Clin. Chim. Acta. 39 (1): 191–9. doi:10.1016/0009-8981(72)90316-6. PMID 4624981.
- ↑ Koorevaar G, Van Stekelenburg GJ (September 1976). "Mammalian acetoacetate decarboxylase activity. Its distribution in subfractions of human albumin and occurrence in various tissues of the rat". Clin. Chim. Acta. 71 (2): 173–83. doi:10.1016/0009-8981(76)90528-3. PMID 963888.
- 1 2 "Human Metabolism" (PDF).
- 1 2 Galassetti PR, Novak B, Nemet D, Rose-Gottron C, Cooper DM, Meinardi S, Newcomb R, Zaldivar F, Blake DR (2005). "Breath ethanol and acetone as indicators of serum glucose levels: an initial report". Diabetes Technol. Ther. 7 (1): 115–23. doi:10.1089/dia.2005.7.115. PMID 15738709.
External links
- acetoacetate decarboxylase at the US National Library of Medicine Medical Subject Headings (MeSH)
- EC 4.1.1.4
- Brenda: Entry of Acetoacetate decarboxylase
- KEGG: Entry of Acetoacetate decarboxylase
- InterPro: IPR010451 Acetoacetate decarboxylase