Gitelman syndrome
| Gitelman syndrome | |
|---|---|
| Classification and external resources | |
| Specialty | endocrinology |
| ICD-10 | N25.8 + E87.6 + E83.4 |
| OMIM | 263800 |
| DiseasesDB | 31860 |
| eMedicine | article/238670 |
| MeSH | D053579 |
| Orphanet | 358 |
Gitelman syndrome is an autosomal recessive kidney disorder characterized by hypokalemic metabolic alkalosis with hypocalciuria, and hypomagnesemia. It is caused by loss of function mutations of the thiazide-sensitive sodium-chloride symporter (also known as NCC, NCCT, or TSC) located in the distal convoluted tubule.[1]
Gitelman syndrome was formerly considered a subset of Bartter syndrome until the distinct genetic and molecular bases of these disorders were identified. Bartter syndrome is also an autosomal recessive hypokalemic metabolic alkalosis, but it derives from a mutation to the NKCC2 found in the thick ascending limb of the loop of Henle.[2]
Cause

Gitelman's syndrome is linked to inactivating mutations in the SLC12A3 gene resulting in a loss of function of the encoded thiazide-sensitive sodium-chloride co-transporter (NCCT). This cell membrane protein participates in the control of ion homeostasis at the distal convoluted tubule portion of the nephron. Loss of this transporter also has the indirect effect of increasing calcium reabsorption in a transcellular fashion. This has been suggested to be the result of a putative basolateral Na-Ca exchanger and apical calcium channel.
When the sodium-chloride cotransporter is inactivated, continued action of the basolateral Na+/K+-ATPase creates a favorable sodium gradient across the basolateral membrane. This increases the reabsorption of divalent cations by secondary active transport. It is currently unknown why calcium reabsorption is increased while magnesium absorption is decreased, leading to hypomagnesemia.
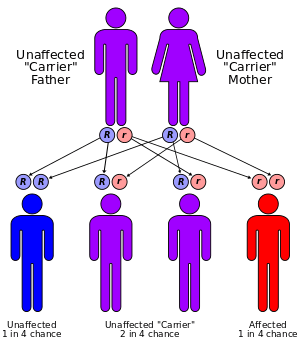
Gitelman's syndrome is an autosomal-recessive disorder: one defective allele has to be inherited from each parent.
Presentation
People suffering from Gitelman's syndrome present symptoms identical to those of patients who are on thiazide diuretics, given that the affected transporter is the exact target of thiazides[3]
Clinical symptoms for this disease are hypochloremic metabolic alkalosis, hypokalemia, and hypocalciuria. Hypomagnesemia is present in many but not all cases. In contrast to patients with Gordon's syndrome, those suffering from Gitelman's syndrome are generally normotensive or hypotensive. Sufferers of Gitelman's syndrome-linked mutations are often complain of severe muscular cramps, numbness or weakness expressed as extreme fatigue or irritability. More severe symptoms such as tetany and paralysis have commonly been reported.Cardiac arrhythmia on ECG and sudden cardiac death and long QT have been reported due to low potassium levels. Phenotypic variations observed among patients probably result from differences in their genetic background and may depend on which particular amino acid in the NCCT protein has been mutated.
See Naesens et al. for a recent review.[4]
Eponym
It is named for Hillel J. Gitelman (1932–2015), an American physician.[5][6][7]
References
- ↑ Simon DB, Nelson-Williams C, Bia MJ, Ellison D, Karet FE, Molina AM, Vaara I, Iwata F, Cushner HM, Koolen M, Gainza FJ, Gitleman HJ, Lifton RP (Jan 1996). "Gitelman's variant of Bartter's syndrome, inherited hypokalaemic alkalosis, is caused by mutations in the thiazide-sensitive Na-Cl cotransporter.". Nat. Genet. 12 (1): 24–30. doi:10.1038/ng0196-24. PMID 8528245.
- ↑ Simon DB, Karet FE, Hamdan JM, DiPietro A, Sanjad SA, Lifton RP (June 1996). "Bartter's syndrome, hypokalaemic alkalosis with hypercalciuria, is caused by mutations in the Na-K-2Cl cotransporter NKCC2". Nat. Genet. 13 (2): 183–8. doi:10.1038/ng0696-183. PMID 8640224.
- ↑ O'Shaughnessy KM, Karet FE (2004). "Salt handling and hypertension". J. Clin. Invest. 113 (8): 1075–81. doi:10.1172/JCI21560. PMC 385413
 . PMID 15085183.
. PMID 15085183. - ↑ Naesens M, Steels P, Verberckmoes R, Vanrenterghem Y, Kuypers D (2004). "Bartter's and Gitelman's syndromes: from gene to clinic". Nephron. Physiology. 96 (3): 65–78. doi:10.1159/000076752. PMID 15056980.
- ↑ synd/2329 at Who Named It?
- ↑ Gitelman HJ, Graham JB, Welt LG (1966). "A new familial disorder characterized by hypokalemia and hypomagnesemia". Trans. Assoc. Am. Physicians. 79: 221–35. PMID 5929460.
- ↑ Unwin RJ, Capasso G (2006). "Bartter's and Gitelman's syndromes: their relationship to the actions of loop and thiazide diuretics" (PDF). CURRENT OPINION IN PHARMACOLOGY. 6 (2): 208–213. doi:10.1016/j.coph.2006.01.002. PMID 16490401.