Discovery and development of integrase inhibitors
The first human immunodeficiency virus (HIV) case was reported in the United States in the early 1980s. Many drugs have been discovered to treat the disease but mutations in the virus and resistance to the drugs make development difficult. Integrase is a viral enzyme that integrates retroviral DNA into the host cell genome. Integrase inhibitors are a new class of drugs used in the treatment of HIV. The first integrase inhibitor, raltegravir, was approved in 2007 and other drugs were in clinical trials in 2011.
History
In the 1980s an infectious disease started to plague human civilization. The coexistence of viruses and humans is a fight for survival for both because the invaders can kill the human but in doing so eliminate their own host. The body uses its immune system to protect itself from bacteria, viruses and other disease-causing beings, and when it fails to do so immunodeficiency diseases occur. One such disease is acquired immunodeficiency syndrome (AIDS) which is most commonly a result of an infection by the human immunodeficiency virus (HIV).[1] Two closely related types of HIV have been identified, HIV-1 and HIV-2. While HIV-2 is spreading in India and West Africa, HIV-1 is more virulent and the number one cause of AIDS worldwide. Though some of the patients have different results in most cases people infected with HIV go on to develop AIDS and ultimately die of opportunistic infections or cancer. Integration to the retroviral genome is critical for gene expression and viral replication. The viral genome is reversely transcribed into the DNA of the infected cell by viral reverse transcriptase, the DNA is then integrated into the host-cell chromosomes with the aid of the viral integrase. RNA transcripts are produced from integrated viral DNA and serve both as mRNAs to direct the synthesis of viral proteins and later as RNA genomes of the new viral particles.Viral particles escape from the cell by budding from the plasma membrane, each enclosed in a membrane envelope.[2] In this process HIV-1 integrase is essential and therefore a very promising target for anti-AIDS drug design. Selective drug design is a possibility as HIV-1 integrase has no known cellular equivalent.[3] Many integrase inhibitors have been discovered and designed but only a few of the molecules were developed further and got as far as phase II or phase III of clinical trials. Raltegravir (brand name Isentress) was granted accelerated approval from the U.S. Food and Drug Administration (FDA) in October 2007 and from EMEA (now EMA) in December 2007.[4][5] It was marketed as an antiretroviral drug (ARV) for HIV-1 infected adults who had already been exposed to a minimum of three ARV classes and showed multi-drug resistance. In general there are two main groups of integrase inhibitors; Integrase Strand Transfer inhibitors (INSTI) and Integrase Binding Inhibitors (INBI). INSTIs restrain the binding of pre-integration complex (PIC) and host DNA and INBIs restrain integrase and viral DNA binding. Raltegravir is an INSTI integrase inhibitor which inhibits both HIV-1 and HIV-2 replication. It is more potent than other previously known integrase inhibitors as well as causing less side effects. Raltegravir, Elvitegravir, and Dolutegravir are the only HIV-1 integrase inhibitor being used to treat HIV infections elvitegravir and S/GSK1349572.[4][6][7][8]
The HIV-1 integrase enzyme
The HIV-1 integrase (IN) is a key enzyme in the replication mechanism of retroviruses.[9] It is responsible for the transfer of virally encoded DNA into the host chromosome which is a necessary event in retroviral replication.[10] Since IN has no equivalent in the host cell, integrase inhibitors have a high therapeutic index as they do not interfere with normal cellular processes.[11]
Structure
IN belongs, both mechanistically and structurally, to the superfamily of polynucleotidyl transferases 10 and is composed of 288 amino acids that form the 32 kDa protein.[9] Retroviruses encode their enzymes (protease, reverse transcriptase and integrase) with the POL gene with the 3´end encoding for IN.[11]

IN is composed of 3 structurally independent, functional domains (see figure 1).:[9]
1. The N-terminal domain (NTD) encompasses amino acids 1–50 and contains two histidine residues (His12 and His16) and two cysteine residues (Cys40 and Cys43), all of which are absolutely conserved and form a HHCC zinc-finger motif.[9][12] Single mutations of any of these four residues reduce IN enzymatic activity.[11] The HHCC zinc-finger motif chelates one zinc atom per IN monomer. The NTD is required for higher order multimer formation which appears to be its primary role.[12][13] The multimerization requires zinc atom that stabilizes the fold.[12]
2. The catalytic core domain (CCD), which encompasses amino acids 51- 212, contains the active site of IN but it can't catalyze integration in the absence of NTD and CTD (the C-terminal domain).[11] CCD contains three absolutely conserved negatively charged amino acids; D64, D116 and E152.[9] These amino acids form the DDE motif that coordinate divalent metal ions (Mg2+ or Mn2+). These metal ions are essential for the catalysis of integration.[12][13] CCD has a mixed β and α structure with five β-sheets and six α helices that are linked by flexible loops.[12] The flexible loops allow conformational changes that are required for 3´processing of the viral DNA and strand transfer (STF) reactions which are two key steps of the integration reaction.[9] CCD is essential for these steps and substitution of any of the residues in the DDE motif dramatically inhibits the activity of IN.[12]
3. The C-terminal domain (CTD), which encompasses amino acids 213–288, binds DNA nonspecifically and its interaction with NTD and CCD is required for IN 3´-processing and strand-transfer activities.[11][12] CTD is the least conserved of the three domains.[12] IN acts as a multimer and dimerization is required for the 3´-processing step, with tetrameric IN catalyzing the strand-transfer reaction.
Function
HIV-1 integration occurs through a multistep process that includes two catalytic reactions: 3´endonucleolytic processing of proviral DNA ends (termed 3´processing) and integration of 3´-processed viral DNA into cellular DNA (referred to as strand transfer).[6] In 3´processing IN binds to a short sequence located at either end of the long terminal repeat (LTR) of the viral DNA and catalyzes endonucleotide cleavage. This results in elimination of a dinucleotide from each of the 3´ends of the LTR. Cleaved DNA is then used as a substrate for integration or strand transfer.[9] Strand transfer is a trans-esterification reaction involving a direct nuleophilic attack of the 3´hydroxy group of the two newly processed viral 3´-DNA ends on the phosphodiester backbone of the host target DNA.[14] This leads to covalent insertion of viral DNA into the genome of the infected cell. Strand transfer occurs simultaneously at both ends of the viral DNA molecule, with an offset of precisely five base pairs between the two opposite points of insertion.[9] The integration reaction is completed by removal of unpaired dinucleotides from the 5'- ends of the viral DNA, repair of the single-stranded gaps created between the viral and target DNA molecules and ligation of 3'-ends to 5'-ends of the host DNA.[9][14] Divalent metals, Mg2+ or Mn2+, are required for 3'-processing and strand transfer steps as well as for assembly of IN onto specific viral donor DNA to form a complex that is competent to carry out either function. Because the abundance of magnesium (Mg2+) versus manganese (Mn 2+) in human cells is 1,000,000-fold, magnesium seems a more readily available divalent cofactor for integration.[6]
Mechanism of action
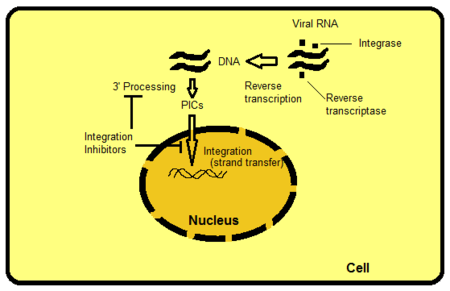
There are several ways to target integrase but strand transfer inhibition is the most intuitively obvious and readily pursued to date. Other targets include, for example, the protein domains beyond the active site of IN. The domains interact with viral or host DNA and are important for binding to the enzyme. It is possible to hamper functions of the enzyme by disrupting or removing these bindings. PIC is a multimeric protein structure inside the host cell, composed of both viral and host proteins. Integrase is a part of PIC‘s viral component. PIC‘s viral and host proteins are believed to modulate intrinsic activity of the enzyme, shuttle PIC to the nucleus and direct integration of viral DNA into a transcriptionally active region of the host genome. If it were possible to exclude certain proteins from the PIC it would block the ability of the virus to integrate into the host genome. The process where the retroviral RNA is transcribed to DNA and then integrated into the host cell's genome is shown in figure 2.[8]
IN strand transfer inhibitors (INSTIs)
Mg2+ and Mn2+ are critical cofactors in the integration phase. Inactivating these cofactors (e.g. through chelation) causes functional impairment of IN. This concept gives researchers the opportunity to design and develop highly efficient IN inhibitors (INIs). In fact, all small molecule HIV-1 INIs that are now being researched contain a structural motif that coordinates the two divalentmagnesium ions in the enzyme’s active site.[6]
Raltegravir and elvitegravir share the same mechanism of action against integrase: to bind to the active site of Mg2+ ions.[8] Competitive inhibitors compete directly with viral DNA for binding to integrase in order to inhibit 3‘-end processing.[15] In doing this the inhibitors completely block the active site from binding to target DNA. This inhibition is called strand transfer inhibition.[8]
Inhibition of the LEDGF/p75- integrase interaction
Lens epithelial derived growth factor (LEDGF/p75) is a host protein that binds to integrase and is crucial for viral replication. The mechanism of action is not precisely known but evidence suggest that LEDGF/p75 guides integrase to insert viral DNA into transcriptionally active sites of the host genome. Inhibitors of this protein are already being developed and patented. They are likely to be highly target specific and less prone to the development of resistance.[8]
IN binding inhibitors
Another class of INIs could be IN binding inhibitors (INBIs) such as V-165. V-165 is a compound shown to inhibit integration but without obvious effect on viral DNA synthesis. When the mechanism of action was studied it showed that V-165 interferes with viral DNA-IN complex formation. Due to its interfering action it is classified as an IN binding inhibitor. Other compounds, such as styrylquinolines share similar mechanism by competing with the LTR substrate for IN binding.[16]
Drug design
Binding
INSTIs bind tightly and specifically to the IN that is associated with the ends of the DNA by chelating the divalent metal ions (Mg2+) which is coordinated by the catalytic triad i.e. the DDE motif.[9] The DDE motif is located in the CCD of IN and is the active site of the enzyme and hence INSTIs are so called active site inhibitors. INSTIs bind to a specific site close to the DDE motif of IN, a site that is present only in the conformation that occurs after processing of the 3´ viral DNA ends. Viral DNA may well form a part of the inhibitor binding site. The binding is a form of allosteric inhibition as it implies blockage of a specific integrase-viral DNA complex.[12] This results in selective inhibition of the strand-transfer reaction, with no significant effect on the 3´-processing reaction.[9] INSTIs may therefore be more specific and bind selectively to the target DNA binding site and hence be less toxic than bifunctional inhibitors that are able to bind to both the donor and target binding sites.[12]
INBIs also bind to IN but the mechanism of action is unknown so the binding can not be detailed.[16]
Structure activity relationship (SAR)
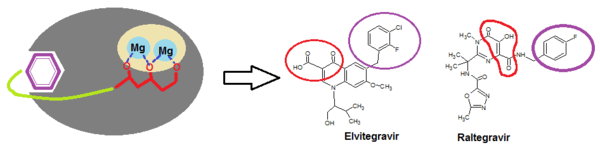
Two structural components are necessary for integrase binding: a hydrophobic benzyl moiety that buries into a highly hydrophobic pocket near the active site; and chelating triad that binds with two Mg2+ ions in a rather hydrophilic region, anchoring the inhibitor onto the protein surface (see figure 3).[17] In fact, all potent integrase inhibitors possess a substituted benzyl component that is critical for maintaining 3‘end joining potency. Removal of the benzyl group prevents inhibitory function.[15] Lipophylic substituents are therefore beneficial for the strand transfer inhibition, in particular the thiophenyl, furanyl and (thiophen-2-yl)phenyl substitutions. Heteroaromatic amine and amide also cause increase in 3‘ processing inhibitory action.[6]
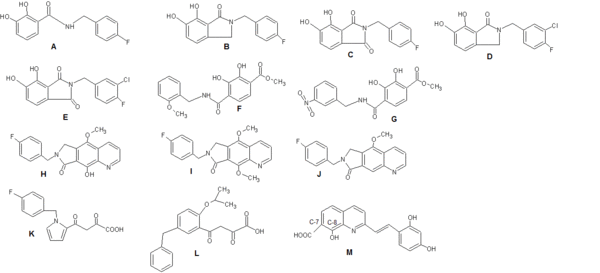
When catechol-based inhibitors of IN were researched it was observed that maintaining a planar relationship with the bis-hydroxylated aryl ring increases potency. The inhibitory activity could be further optimized by including a meta-chloro substituent, enhancing the interaction of the benzyl group with the adjacent hydrophobic pocket (see figure 4: Structures A-G).[8]
A benzyl substituted hydroxyl group (fig. 4 H) improves metal-chelating capability (compared to structure J in fig. 4) while a methoxy group (I) is much less potent due to steric clash by the additional methyl group with the catalytic metals.[15]
When researching diketo derivates, the central pyrrole ring of structure K in fig. 4 was replaced by a series of aromatic systems having various substitution patterns. That provided optimum relative orientation of the benzyl and diketoacid (DKA) site chain. Structure L in fig. 4 resulted in 100 fold increase in potency.[18]
Benard et al (2004) synthesized INIs with a quinoline subunit and an ancillary aromatic ring linked by functionalized spacers such as amide, hydrazide, urea and hydroxyprop-1-en-3-one moiety. They found that the amide group containing dervates were the most promising ones.[18][19] By synthesizing series of styrylquinones researchers found out that a carboxyl group at C-7, a hydroxylgroup at C-8 in the quinoline subunit and an ancillary phenyl ring (Figure 4: Structure M) are required for inhibition, although alterations of the ring are tolerated. Two hydroxyl groups on the ancillary phenyl ring are also required for inhibitory potency.[18]
Pharmacophore
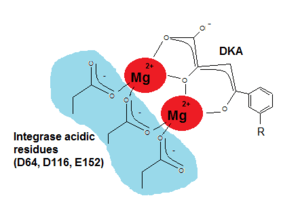
Since critical structure information is scarce on HIV integrase catalysis it is difficult to find the exact pharmacophore for its inhibition. Wang et al (2010) hoped that by studying the SAR and pharmacophore of a dual inhibitor scaffold, focusing both on integrase and reverse transcriptase (RT) it would be possible to observe anti-integrase activity. By studying the SAR of HIV integrase inhibitors it was possible to find that for optimal integrase inhibition the pharmacophore requires a regiospecific (N-1) DKA of a specific length. A DKA functionality or its heterocyclic bioisostere that selectively inhibit strand transfer seem to be present in all major chemotypes of integrase inhibitors.[17] As detailed in the SAR discussion above the two necessary structural components of INI are a benzyl hydrophobic moiety and a chelating triad to bind the Mg2+ ions. For the triad to bind the Mg2+ ions has to be ionized (see fig. 5) and thus a pharmacophore bioisostere has to be ionized too and the benzyl pharmacophore bioisostere must to be very hydrophobic.[11][17]
However, despite previous success in clinical development (raltegravir), a detailed binding model is lacking so it has proven difficult to structure base the design of integrase inhibitors. When the pharmacophore of salicylic acid and catechol were merged, new chemical scaffolds were created. The adjacent hydroxyl and carboxylic groups on salicylic acid could bind with the metal ions and serve as their pharmacophore. Polyhydroxylated aromatic inhibitors are mostly active against strand transfer reactions and 3‘-processing which suggests a mechanism that targets both steps. This is a very important part of the compound as it can be used to bind to the divalent metal on the active site of IN and as such be effective against viral strains that are resistant to strand transfer specific inhibitors.[6][17]
Resistance
It has been discovered that over 60 variations of INSTI mutations cause in vivo and in vitro resistance. Due to these mutations and development of resistance the inhibitors are less effective against the virus.[9] Resistance of INI corresponds to those of other ARV drugs. First IN resistance is caused by primary mutations that decrease INI sensitivity in combination with secondary mutations that further reduce virus sensitivity and/or repair decreased fitness of the virus. Secondly there is a genetic barrier to INI resistance, defined by the number of mutations required for the loss of clinical INI activity. Thirdly there is extensive but incomplete cross-resistance among the INIs.[13] A loop containing amino acid residues 140–149 is located in the catalytic-core domain and is important for IN function as mentioned before. This loop is flexible and even though its role is not quite known it is thought to be important and its functions critical for DNA binding. This resistance appears within mutations in this IN-coding region.[9] The resistance to raltegravir and elvitegravir is primarily due to the same two mutation pathways but other primary mutations are also involved for each of the drugs.[10] Some mutations increase resistance to the drugs to a large extent than others. For example, one of the most common mutation pathway increases the resistance to raltegravir up to 100 times more than the second most common one.[9] Resistance to Integrase Inhibitor S/GSK1349572 is still being developed and the resistance has not been fully characterized. When it was assessed alongside the primary mutations of raltegravir and elvitegravir it did not show cross-resistance which means that it could be useful against drug resistant viruses.[7] Raltegravir has limited intestinal absorption and thus resistance cannot be overcome by prescribing higher doses. Newer drugs are warranted to overcome this pharmacological disadvantage and gain plasma concentrations high enough to target raltegravir-resistant viruses.[7]
Current status

The search for new ways to improve treatment of patients infected with HIV is constant. Considering the experience that has been gathered since the 1980s of ARV drug development arrival of INSTIs as a new potent class of ARV signals a new era in the treatment of HIV. Development of a successful INSTI treatment was accomplished when raltegravir was discovered by Merck Sharp & Dohme Limited.[12] A conditional marketing authorization was licensed in December 2007 by the European Commission which was valid throughout the European Union.[20] In 2009 this authorization was converted to a full marketing authorization and in the same year the FDA changed the approval from accelerated to traditional approval and listed the drug as a first line ARV treatment agent.[12][21] The second INSTI drug, elvitegravir, was identified by Japan Tobacco and clinical trials began in 2005. In 2011 the drug was still in phase three clinical trials, where it is being compared to raltegravir, in treatment experienced subjects and is also in phase two development in naïve subjects as a part of a multidrug treatment.[12] S/GSK1349572 is an integrase inhibitor discovered by ViiV/Shinongi which was entering phase three in clinical trials in 2011. This new drug is promising and seems to be well tolerated and so far shows better results than both raltegravir and elvitegravir.[22]
Since there have been problems with resistance to raltegravir and elvitegravir, scientists have started to work on new second generation integrase inhibitors, such as MK-2048 which in 2009 was developed by Merck. It's a prototype second generation INSTI that remains potent against viruses containing mutations against raltegravir and elvitegravir. The mechanism of action and SAR of MK-2048 is the same as of the other INSTIs, the structure of MK-2048 shown in figure 6 with essential pharmacophore highlighted.[23][24]
Even though drugs discussed above are promising the development has a long way to go and many things are still unknown about the efficacy, safety and mechanism of action of these drugs.[7]
See also
- Raltegravir
- Elvitegravir
- Integrase
- Integrase inhibitor
- HIV
- Reverse transcriptase
- MK-2048
- Dolutegravir
References
- ↑ Johnson, Dee Unglaub Silverthorn ; with William C. Ober, illustration coordinator ; Claire W. Garrison, illustrator ; Andrew C. Silverthorn, clinical consultant ; with contributions by Bruce R. (2007). Human physiology : an integrated approach (4th ed.). San Francisco: Pearson/Benjamin Cummings. ISBN 0-8053-6849-3.
- ↑ Murphy, Kenneth; Travers, Paul; Walport, Mark (2008). Janeway's immunobiology (7th ed.). New York: Garland Science. ISBN 978-0-8153-4123-9.
- ↑ Pommier, Y; Marchand, C; Neamati, N (Sep 2000). "Retroviral integrase inhibitors year 2000: update and perspectives". Antiviral Research. 47 (3): 139–48. doi:10.1016/S0166-3542(00)00112-1. PMID 10974366.
- 1 2 Dąbrowska, Magdalena Monika; Wiercińska-Drapało, Alicja (1 Jan 2007). "Integrase inhibitors as a new class of ARV treatment". HIV & AIDS Review. 6 (4): 10–14. doi:10.1016/S1730-1270(10)60053-7.
- ↑ "FDA approval of Isentress (raltegravir)". U.S. Food and Drug Administration (FDA). Retrieved 25 Sep 2011.
- 1 2 3 4 5 6 Fan, X; Zhang, FH; Al-Safi, RI; Zeng, LF; Shabaik, Y; Debnath, B; Sanchez, TW; Odde, S; Neamati, N; Long, YQ (2011-08-15). "Design of HIV-1 integrase inhibitors targeting the catalytic domain as well as its interaction with LEDGF/p75: a scaffold hopping approach using salicylate and catechol groups". Bioorganic & Medicinal Chemistry. 19 (16): 4935–52. doi:10.1016/j.bmc.2011.06.058. PMC 3163123
 . PMID 21778063.
. PMID 21778063. - 1 2 3 4 Lenz, JC; Rockstroh, JK (Apr 2011). "S/GSK1349572, a new integrase inhibitor for the treatment of HIV: promises and challenges". Expert Opinion on Investigational Drugs. 20 (4): 537–48. doi:10.1517/13543784.2011.562189. PMID 21381981.
- 1 2 3 4 5 6 Pendri, A; Meanwell, NA; Peese, KM; Walker, MA (Aug 2011). "New first and second generation inhibitors of human immunodeficiency virus-1 integrase". Expert opinion on therapeutic patents. 21 (8): 1173–89. doi:10.1517/13543776.2011.586631. PMID 21599420.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 Mouscadet, JF; Delelis, O; Marcelin, AG; Tchertanov, L (Aug–Oct 2010). "Resistance to HIV-1 integrase inhibitors: A structural perspective". Drug resistance updates : reviews and commentaries in antimicrobial and anticancer chemotherapy. 13 (4–5): 139–50. doi:10.1016/j.drup.2010.05.001. PMID 20570551.
- 1 2 Cocohoba, J; Dong, BJ (Oct 2008). "Raltegravir: the first HIV integrase inhibitor". Clinical therapeutics. 30 (10): 1747–65. doi:10.1016/j.clinthera.2008.10.012. PMID 19014832.
- 1 2 3 4 5 6 Pommier, Yves; Johnson, Allison A.; Marchand, Christophe (24 Feb 2005). "Integrase inhibitors to treat HIV/Aids". Nature Reviews Drug Discovery. 4 (3): 236–248. doi:10.1038/nrd1660. PMID 15729361.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 McColl, DJ; Chen, X (Jan 2010). "Strand transfer inhibitors of HIV-1 integrase: bringing IN a new era of antiretroviral therapy". Antiviral Research. 85 (1): 101–18. doi:10.1016/j.antiviral.2009.11.004. PMID 19925830.
- 1 2 3 Blanco, JL; Varghese, V; Rhee, SY; Gatell, JM; Shafer, RW (2011-05-01). "HIV-1 integrase inhibitor resistance and its clinical implications". The Journal of Infectious Diseases. 203 (9): 1204–14. doi:10.1093/infdis/jir025. PMC 3069732. PMID 21459813.
- 1 2 De Luca, Laura; De Grazia, Sara; Ferro, Stefania; Gitto, Rosaria; Christ, Frauke; Debyser, Zeger; Chimirri, Alba (Feb 2011). "HIV-1 integrase strand-transfer inhibitors: design, synthesis and molecular modeling investigation". European journal of medicinal chemistry. 46 (2): 756–764. doi:10.1016/j.ejmech.2010.12.012. PMID 21227550.
- 1 2 3 Chen, X; Tsiang, M; Yu, F; Hung, M; Jones, GS; Zeynalzadegan, A; Qi, X; Jin, H; Kim, CU; Swaminathan, S; Chen, JM (2008-07-11). "Modeling, analysis, and validation of a novel HIV integrase structure provide insights into the binding modes of potent integrase inhibitors". Journal of Molecular Biology. 380 (3): 504–19. doi:10.1016/j.jmb.2008.04.054. PMID 18565342.
- 1 2 Hombrouck, A.; Hantson, A.; van Remoortel, B.; Michiels, M.; Vercammen, J.; Rhodes, D.; Tetz, V.; Engelborghs, Y.; Christ, F.; Debyser, Z.; Witvrouw, M. (Jun 2007). "Selection of human immunodeficiency virus type 1 resistance against the pyranodipyrimidine V-165 points to a multimodal mechanism of action". The Journal of antimicrobial chemotherapy. 59 (6): 1084–95. doi:10.1093/jac/dkm101. PMID 17470918.
- 1 2 3 4 Wang, Z; Tang, J; Salomon, CE; Dreis, CD; Vince, R (2010-06-15). "Pharmacophore and structure-activity relationships of integrase inhibition within a dual inhibitor scaffold of HIV reverse transcriptase and integrase". Bioorganic & Medicinal Chemistry. 18 (12): 4202–11. doi:10.1016/j.bmc.2010.05.004. PMID 20576573.
- 1 2 3 Dubey, S; Satyanarayana, YD; Lavania, H (Sep 2007). "Development of integrase inhibitors for treatment of AIDS: an overview". European journal of medicinal chemistry. 42 (9): 1159–68. doi:10.1016/j.ejmech.2007.01.024. PMID 17367896.
- ↑ Bénard, C; Zouhiri, F; Normand-Bayle, M; Danet, M; Desmaële, D; Leh, H; Mouscadet, JF; Mbemba, G; Thomas, CM; Bonnenfant, S; Le Bret, M; d'Angelo, J (2004-05-17). "Linker-modified quinoline derivatives targeting HIV-1 integrase: synthesis and biological activity". Bioorganic & Medicinal Chemistry Letters. 14 (10): 2473–6. doi:10.1016/j.bmcl.2004.03.005. PMID 15109635.
- ↑ "Isentress" (PDF). European Medicines Agency. Retrieved 17 Sep 2011.
- ↑ "Traditional approval of Isentress (raltegravir)". U.S. Food and Drug Administration (FDA). Retrieved 25 Sep 2011.
- ↑ Barnhart, Matthew; James Shelton (April 2011). "A better state of ART improving antiretroviral regimens to increase global access to HIV treatment". Journal of AIDS and HIV Research. 3 (4): 71–78.
- ↑ Bar-Magen, T; Sloan, RD; Donahue, DA; Kuhl, BD; Zabeida, A; Xu, H; Oliveira, M; Hazuda, DJ; Wainberg, MA (Sep 2010). "Identification of novel mutations responsible for resistance to MK-2048, a second-generation HIV-1 integrase inhibitor". Journal of Virology. 84 (18): 9210–6. doi:10.1128/JVI.01164-10. PMC 2937597. PMID 20610719.
- ↑ Goethals, O; Vos, A,; Van Ginderen, M; Geluykens, P; Smits, V; Schols, D; Hertogs, K; Clayton, R (2010-07-05). "Primary mutations selected in vitro with raltegravir confer large fold changes in susceptibility to first-generation integrase inhibitors, but minor fold changes to inhibitors with second-generation resistance profiles". Virology. 402 (2): 338–46. doi:10.1016/j.virol.2010.03.034. PMID 20421122.