Velvet assembler
| Developer(s) |
|
|---|---|
| Initial release | 2008 |
| Stable release |
1.2.10
|
| Development status | Active |
| Operating system | UNIX-based |
| Available in | C |
| Type | Bioinformatics |
| License | GPL |
| Website | http://www.ebi.ac.uk/~zerbino/velvet/ |
Velvet is an algorithm package that has been designed to deal with de novo genome assembly and short read sequencing alignments. This is achieved through the manipulation of de Bruijn graphs for genomic sequence assembly via the removal of errors and the simplification of repeated regions.[2] Velvet has also been implemented inside of commercial packages, such as Sequencher, Geneious, MacVector and BioNumerics.
Introduction
The development of next-generation sequencers (NGS) allowed for increased cost effectiveness on very short read sequencing. The manipulation of de Bruijn graphs as a method for alignment became more realistic but further developments were needed to address issues with errors and repeats.[3] This led to the development of Velvet by Daniel Zerbino and Ewan Birney at the European Bioinformatics Institute in the United Kingdom.[4]
Velvet works by efficiently manipulating de Bruijn graphs through simplification and compression, without the loss of graph information, by converging non-intersecting paths into single nodes. It eliminates errors and resolves repeats by first using an error correction algorithm that merges sequences together. Repeats are then removed from the sequence via the repeat solver that separates paths which share local overlaps.
The combination of short reads and read pairs allows Velvet to resolve small repeats and produce contigs of reasonable length. This application of Velvet can produce contigs with a N50 length of 50 kb on paired-end prokaryotic data and a 3 kb length for regions of mammalian data.
Algorithm
As already mentioned Velvet uses the de Bruijn graph to assemble short reads. More specifically Velvet represents each different k-mer obtained from the reads by a unique node on the graph. Two nodes are connected if its k-mers have a k-1 overlap. In other words, an arc from node A to node B exists if the last k-1 characters of the k-mer, represented by A, are the first k-1 characters of the k-mer represented by B. The following figure shows an example of a de Bruijn graph generated with Velvet:
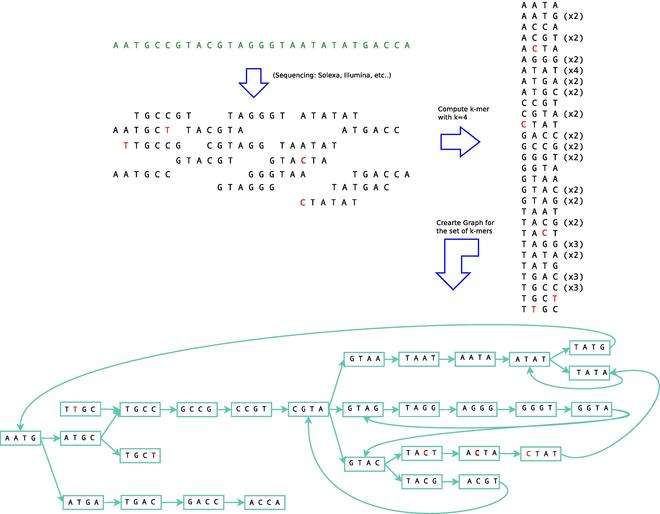
The same process is simultaneously done with the reverse complement of all the k-mers to take into account the overlaps between the reads of opposite strands. A number of optimizations can be done over the graph which includes simplification and error removal.
Simplification
An easy way to save memory costs is to merge nodes that do not affect the path generated in the graph, i.e., whenever a node A has only one outgoing arc that points to node B, with only one ingoing arc, the nodes can be merged. It is possible to represent both nodes as one, merging them and all their information together. The next figure illustrates this process in the simplification of the initial example.
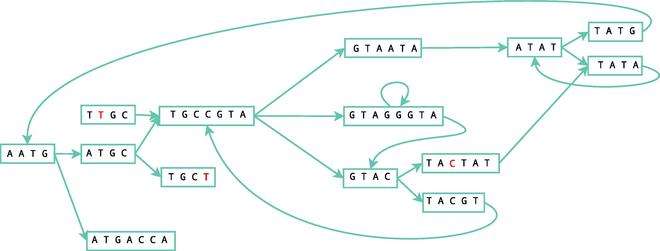
Error removal
Errors in the graph can be caused by the sequencing process or it could simply be that the biological sample contains some errors (for example polymorphisms). Velvet recognizes three kinds of errors: tips; bubbles; and erroneous connections.
Tips
A node is considered a tip and should be erased if it is disconnected on one of its ends, the length of the information stored in the node is shorter than 2k, and the arc leading to this node has a low multiplicity (number of times the arc was found during the construction of the graph) and as a result cannot be compared to other alternative paths. Once these errors are removed, the graph once again undergoes simplification.
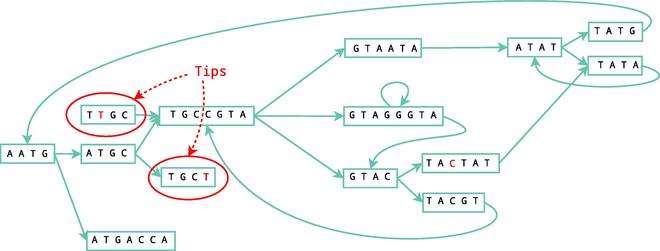
Bubbles
Bubbles are generated when two paths start and end at the same nodes. Normally bubbles are caused by errors or biological variants. These errors are removed using the Tour Bus algorithm, which is similar to a Dijkstra's algorithm, a breadth-first search that detects the best path to follow and determines which ones should be erased. A simple example is shown in figure 4.

This process is also shown in figure 5 following on from the examples shown in figures 1 and 2.
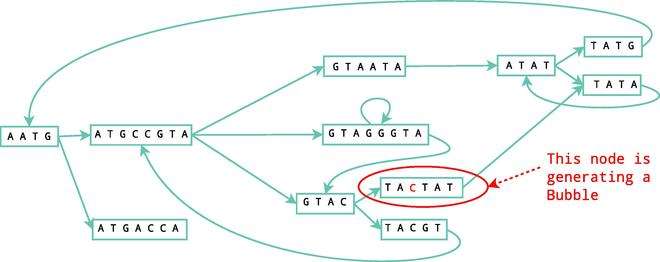
Erroneous connections
These are connections that do not generate correct paths or do not create any recognizable structures within the graph. Velvet erases these errors after completion of the Tour Bus algorithm, applying a simple coverage cut-off that must be defined by the user.
Velvet commands
Velvet provides the following functions:
- velveth
- This command helps to construct the data set (hashes the reads) for velvetg and includes information about the meaning of each sequence files.
- velvetg
- This command builds the de Bruijn graph from the k-mers obtained by velveth and runs simplification and error correction over the graph. It then extracts the contigs.
After running velvetg a number of files are generated. Most importantly, a file of contigs contains the sequences of the contigs longer than 2k, where k is the word-length used in velveth.
For more detail and examples refer to the Velvet Manual [5]
Motivation
Current DNA sequencing technologies, including NGS, are limited on the basis that genomes are much larger than any read length. Typically, NGS operate with small reads, less than 400 bp, and have a much lower cost per read than previous first generation machines. They are also simpler to operate with higher parallel operation and higher yield.[3]
However, short reads contain less information than larger reads thus requiring a higher assembly read coverage to allow for detectable overlaps. This in turn increases the complexity of the sequencing and significantly increases computational requirements. A larger number of reads also increases the size of the overlap graph, making it more difficult and lengthy to compute. The connections between the reads become more indistinct due to the decrease in overlapping sections leading to a greater possibility of errors.
To overcome these issues, dynamic sequencing programs that are efficient, highly cost effective and able to resolve errors and repeats were developed. Velvet algorithms was designed for this and are able to perform short read de novo sequencing alignment in relatively short computational time and with lower memory usage compared to other assemblers.[6]
Graphical interface
One of the main drawbacks in the use of Velvet is the use of the command-line interface and the difficulties users, especially beginners, face in the implementation of their data.[7] It was developed in 2012 and designed to overcome this problem and simplify the running of Velvet.
See also
References
- ↑ Zerbino, D. R. (2010). "Using the Velvetde novo Assembler for Short-Read Sequencing Technologies". In Andreas D. Baxevanis. Using the Velvet de novo assembler for short-read sequencing technologies. pp. Unit 11.5. doi:10.1002/0471250953.bi1105s31. ISBN 0471250953. PMC 2952100
 . PMID 20836074.
. PMID 20836074. - ↑ Zerbino, D. R.; Birney, E. (2008). “Velvet: de novo assembly using very short reads”. Retrieved 2013-10-18.
- 1 2 Miller, J. R.; Koren, S; Sutton, G (2010). "Assembly algorithms for next-generation sequencing data". Genomics. 95 (6): 315–27. doi:10.1016/j.ygeno.2010.03.001. PMC 2874646. PMID 20211242.
- ↑ Zerbino, D. R.; Birney, E. (2008). "Velvet: Algorithms for de novo short read assembly using de Bruijn graphs". Genome Research. 18 (5): 821–829. doi:10.1101/gr.074492.107. PMC 2336801. PMID 18349386.
- ↑ “Velvet Manual” Retrieved 2013-10-18
- ↑ Zhang, W.; Chen, J.; Yang, Y.; Tang, Y.; Shang, J.; Shen, B. (2011). "A Practical Comparison of De Novo Genome Assembly Software Tools for Next-Generation Sequencing Technologies". PLoS ONE. 6 (3): e17915. doi:10.1371/journal.pone.0017915. PMC 3056720. PMID 21423806.
- ↑ Powell, D. R.; Seemann, T (2013). "VAGUE: A graphical user interface for the Velvet assembler". Bioinformatics. 29 (2): 264–5. doi:10.1093/bioinformatics/bts664. PMID 23162059.