System size expansion
The system size expansion, also known as van Kampen's expansion or the Ω-expansion, is a technique pioneered by Nico van Kampen[1] used in the analysis of stochastic processes. Specifically, it allows one to find an approximation to the solution of a master equation with nonlinear transition rates. The leading order term of the expansion is given by the linear noise approximation, in which the master equation is approximated by a Fokker–Planck equation with linear coefficients determined by the transition rates and stoichiometry of the system.
Less formally, it is normally straightforward to write down a mathematical description of a system where processes happen randomly (for example, radioactive atoms randomly decay in a physical system, or genes that are expressed stochastically in a cell). However, these mathematical descriptions are often too difficult to solve for the study of the systems statistics (for example, the mean and variance of the number of atoms or proteins as a function of time). The system size expansion allows one to obtain an approximate statistical description that can be solved much more easily than the master equation.
Preliminaries
Systems that admit a treatment with the system size expansion may be described by a probability distribution  , giving the probability of observing the system in state
, giving the probability of observing the system in state 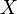 at time
at time 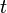 . may be, for example, a vector with elements corresponding to the number of molecules of different chemical species in a system. In a system of size
. may be, for example, a vector with elements corresponding to the number of molecules of different chemical species in a system. In a system of size 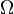 (intuitively interpreted as the volume), we will adopt the following nomenclature:
(intuitively interpreted as the volume), we will adopt the following nomenclature: 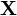 is a vector of macroscopic copy numbers,
is a vector of macroscopic copy numbers, 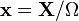 is a vector of concentrations, and
is a vector of concentrations, and 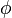 is a vector of deterministic concentrations, as they would appear according to the rate equation in an infinite system.
is a vector of deterministic concentrations, as they would appear according to the rate equation in an infinite system. 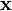 and are thus quantities subject to stochastic effects.
and are thus quantities subject to stochastic effects.
A master equation describes the time evolution of this probability.[1] Henceforth, a system of chemical reactions[2] will be discussed to provide a concrete example, although the nomenclature of "species" and "reactions" is generalisable. A system involving 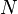 species and
species and 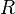 reactions can be described with the master equation:
reactions can be described with the master equation:

Here, is the system size, 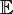 is an operator which will be addressed later,
is an operator which will be addressed later, 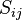 is the stoichiometric matrix for the system (in which element gives the stoichiometric coefficient for species
is the stoichiometric matrix for the system (in which element gives the stoichiometric coefficient for species  in reaction
in reaction 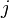 ), and
), and  is the rate of reaction given a state and system size .
is the rate of reaction given a state and system size .
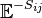 is a step operator,[1] removing from the th element of its argument. For example,
is a step operator,[1] removing from the th element of its argument. For example, 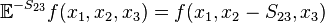 . This formalism will be useful later.
. This formalism will be useful later.
The above equation can be interpreted as follows. The initial sum on the RHS is over all reactions. For each reaction , the brackets immediately following the sum give two terms. The term with the simple coefficient −1 gives the probability flux away from a given state due to reaction changing the state. The term preceded by the product of step operators gives the probability flux due to reaction changing a different state 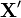 into state . The product of step operators constructs this state .
into state . The product of step operators constructs this state .
Example
For example, consider the (linear) chemical system involving two chemical species 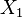 and
and  and the reaction
and the reaction 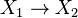 . In this system,
. In this system,  (species),
(species), 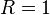 (reactions). A state of the system is a vector
(reactions). A state of the system is a vector 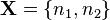 , where
, where 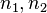 are the number of molecules of and respectively. Let
are the number of molecules of and respectively. Let 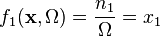 , so that the rate of reaction 1 (the only reaction) depends on the concentration of . The stoichiometry matrix is
, so that the rate of reaction 1 (the only reaction) depends on the concentration of . The stoichiometry matrix is  .
.
Then the master equation reads:

where 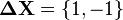 is the shift caused by the action of the product of step operators, required to change state to a precursor state
is the shift caused by the action of the product of step operators, required to change state to a precursor state  .
.
Linear noise approximation
If the master equation possesses nonlinear transition rates, it may be impossible to solve it analytically. The system size expansion utilises the ansatz that the variance of the steady-state probability distribution of constituent numbers in a population scales like the system size. This ansatz is used to expand the master equation in terms of a small parameter given by the inverse system size.
Specifically, let us write the 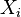 , the copy number of component , as a sum of its "deterministic" value (a scaled-up concentration) and a random variable
, the copy number of component , as a sum of its "deterministic" value (a scaled-up concentration) and a random variable 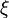 , scaled by
, scaled by 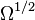 :
:
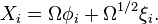
The probability distribution of can then be rewritten in the vector of random variables :

Let us consider how to write reaction rates  and the step operator in terms of this new random variable. Taylor expansion of the transition rates gives:
and the step operator in terms of this new random variable. Taylor expansion of the transition rates gives:
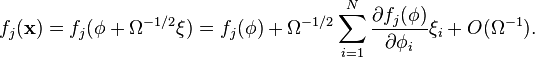
The step operator has the effect 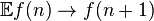 and hence
and hence 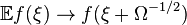 :
:

We are now in a position to recast the master equation.
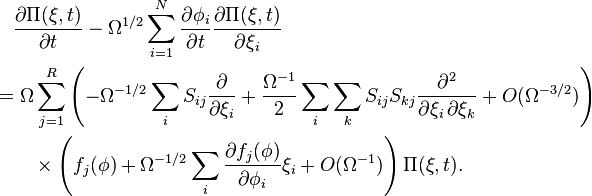
This rather frightening expression makes a bit more sense when we gather terms in different powers of . First, terms of order give
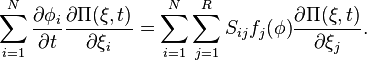
These terms cancel, due to the macroscopic reaction equation
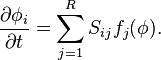
The terms of order 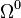 are more interesting:
are more interesting:
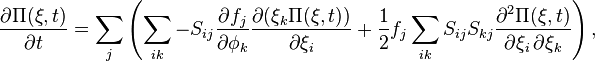
which can be written as
![\frac{\partial \Pi (\mathbf{\xi}, t)}{\partial t} = -\sum_{ik} A_{ik} \frac{\partial (\xi_k \Pi)}{\partial \xi_i} + \frac{1}{2} \sum_{ik} [\mathbf{BB}^T]_{ik} \frac{\partial^2 \Pi}{\partial \xi_i \, \partial \xi_k},](../I/m/4d99879f78d613afc8b51a88f25c8933.png)
where

and
![[ \mathbf{BB}^T ]_{ik} = \sum_{j = 1}^R S_{ij}S_{kj} f_j (\mathbf{\phi}) = [ \mathbf{S} \, \mbox{diag}(f(\mathbf{\phi})) \, \mathbf{S}^T ]_{ik}.](../I/m/593d4743162c22c360d2561c784d758f.png)
The time evolution of 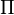 is then governed by the linear Fokker–Planck equation with coefficient matrices
is then governed by the linear Fokker–Planck equation with coefficient matrices 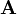 and
and  (in the large- limit, terms of
(in the large- limit, terms of 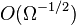 may be neglected, termed the linear noise approximation). With knowledge of the reaction rates
may be neglected, termed the linear noise approximation). With knowledge of the reaction rates 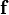 and stoichiometry
and stoichiometry 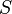 , the moments of can then be calculated.
, the moments of can then be calculated.
Software
The linear noise approximation has become a popular technique for estimating the size of intrinsic noise in terms of coefficients of variation and Fano factors for molecular species in intracellular pathways. The second moment obtained from the linear noise approximation (on which the noise measures are based) are exact only if the pathway is composed of first-order reactions. However bimolecular reactions such as enzyme-substrate, protein-protein and protein-DNA interactions are ubiquitous elements of all known pathways; for such cases, the linear noise approximation can give estimates which are accurate in the limit of large reaction volumes. Since this limit is taken at constant concentrations, it follows that the linear noise approximation gives accurate results in the limit of large molecule numbers and becomes less reliable for pathways characterized by many species with low copy numbers of molecules.
A number of studies have elucidated cases of the insufficiency of the linear noise approximation in biological contexts by comparison of its predictions with those of stochastic simulations.[3][4] This has led to the investigation of higher order terms of the system size expansion that go beyond the linear approximation. These terms have been used to obtain more accurate moment estimates for the mean concentrations and for the variances of the concentration fluctuations in intracellular pathways. In particular, the leading order corrections to the linear noise approximation yield corrections of the conventional rate equations.[5] Terms of higher order have also been used to obtain corrections to the variances and covariances estimates of the linear noise approximation.[6][7] The linear noise approximation and corrections to it can be computed using the open source software intrinsic Noise Analyzer. The corrections have been shown to be particularly considerable for allosteric and non-allosteric enzyme-mediated reactions in intracellular compartments.
References
- 1 2 3 van Kampen, N. G. (2007) "Stochastic Processes in Physics and Chemistry", North-Holland Personal Library
- ↑ Elf, J. and Ehrenberg, M. (2003) "Fast Evaluation of Fluctuations in Biochemical Networks With the Linear Noise Approximation", Genome Research, 13:2475–2484.
- ↑ Hayot, F. and Jayaprakash, C. (2004), "The linear noise approximation for molecular fluctuations within cells", Physical Biology, 1:205
- ↑ Ferm, L. Lötstedt, P. and Hellander, A. (2008), "A Hierarchy of Approximations of the Master Equation Scaled by a Size Parameter", Journal of Scientific Computing, 34:127
- ↑ Grima, R. (2010) "An effective rate equation approach to reaction kinetics in small volumes: Theory and application to biochemical reactions in nonequilibrium steady-state conditions", The Journal of Chemical Physics, 132:035101
- ↑ Grima, R. and Thomas, P. and Straube, A.V. (2011), "How accurate are the nonlinear chemical Fokker-Planck and chemical Langevin equations?", The Journal of Chemical Physics, 135:084103
- ↑ Grima, R. (2012), "A study of the accuracy of moment-closure approximations for stochastic chemical kinetics", The Journal of Chemical Physics, 136: 154105