Synaptic fatigue
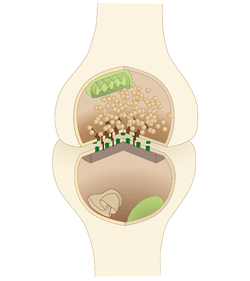
Synaptic fatigue, or short-term synaptic depression, is an activity-dependent form of short term synaptic plasticity that results in the temporary inability of neurons to fire and therefore transmit an input signal. It is thought to be a form of negative feedback in order to physiologically control particular forms of nervous system activity.[1]
It is caused by a temporary depletion of synaptic vesicles that house neurotransmitters in the synapse, generally produced by persistent high frequency neuronal stimulation. The neurotransmitters are released by the synapse to propagate the signal to the postsynaptic cell. It has also been hypothesized that synaptic fatigue could be a result of postsynaptic receptor desensitization or changes in postsynaptic passive conductance, but recent evidence has suggested that it is primarily a presynaptic phenomenon.[2][3]
Background
Chemical synapses allow for signal transmission by a presynaptic cell releasing neurotransmitters into the synapse to bind to receptors on a postsynaptic cell. These neurotransmitters are synthesized in the presynaptic cell and housed in vesicles until released. Once neurotransmitters are released into the synaptic cleft and a signal is relayed, re-uptake begins which is the process of transport proteins clearing out the neurotransmitters from the synapse and recycling them in order to allow for a new signal to be propagated. If stimulation is occurring at a high enough frequency and with enough strength, neurotransmitters will be released at a faster rate than re-uptake can recycle them which will ultimately deplete them until there are no longer readily releasable vesicles and a signal can no longer be transmitted.
Functional significance
It has previously been shown that repeated short trains of action potentials causes an exponential decay of the synaptic response amplitudes in the neurons of many neural networks, specifically the caudal pontine reticular nucleus (PnC). Recent research has suggested that only repeated burst stimulation, as opposed to single or paired pulse stimulation, at a very high frequency can result in SF.[2] Some cells like aortic baroreceptor neurons could have devastating effects including the inability to regulate aortic blood pressure if the onset of synaptic fatigue were to affect them. Metabotropic glutamate autoreceptor activation in these neurons may inhibit synaptic transmission by inhibiting calcium influx, decreasing synaptic vesicle exocytosis and modulating the mechanisms governing synaptic vesicle recovery and endocytosis.[4] These glutamate autoreceptors are able to inhibit synaptic fatigue in order to prevent the detrimental physiological consequences that could result from dysfunctional blood pressure regulation in the aorta (not true)
Synaptic recovery
When synaptic vesicles release neurotransmitters into the synapse that bind with post-synaptic membrane proteins to pass a signal, neurotransmitter re-uptake occurs to recycle neurotransmitters in the presynaptic cell in order to be released again. Neurotransmitter vesicles are recycled through the process of endocytosis. Because each presynaptic cell can link up to thousands of connections with other neurons, synaptic fatigue and its recovery can cause interactions with other neuronal circuits and can affect the kinetics with other processes of neurons.[5] It is important that the recycling of neurotransmitters take place at an effective and efficient rate in order to prevent synaptic fatigue from negatively affecting signal transmission.
Timing
Maintaining a readily releasable vesicle pool is important in allowing for the constant ability to pass physiological signals between neurons. The timing it takes for neurotransmitter to be released into the synaptic cleft and then be recycled back to the presynaptic cell to be reused is not currently well understood. There are two models currently proposed to attempt to understand this process. One model predicts that the vesicle undergoes complete fusion with the presynaptic cellular membrane once all its contents have been emptied. It then must retrieve vesicular membrane from other sites which could take up to tens of seconds.[6] The second model tries to explain this phenomenon by assuming the vesicles immediately begin to recycle neurotransmitters after release, which takes less than a second to complete endocytosis.[6] One study showed varying times of complete endocytosis ranging from 5.5-38.9 seconds. It also indicated that these times were completely independent of long term or chronic activity.[7]
Affected cells
Synaptic fatigue can affect many synapses of many different types of neurons.[5] The existence and observations of synaptic fatigue are accepted universally, although the exact mechanisms underlying the phenomenon are not completely understood. It is generally seen in mature cells at high frequencies of stimuli (>1 Hz). One specific example is that the gill withdrawal reflex of the Aplysia is caused by homosynaptic depression.[8] Although homosynaptic and heterosynaptic depression can lead to long-term depression and/or potentiation, this particular case is a short-term example of how homosynaptic depression causes synaptic fatigue. Perforant path–granule cells (PP-GC) in the dentate gyrus of the hippocampus in adult rats have been shown to experience fatigue at lower frequencies (0.05-0.2 Hz).[9] In the developing rat PP-GCs, two types of synaptic plasticity were shown to lead to synaptic fatigue. A low frequency reversible depression of presynaptic vesicle release and a form of nonreversible depression caused by AMPA silencing. The second form of plasticity disappears with maturation of PP-GCs, although the reversible low frequency depression remains unchanged.[9]
Role in neural plasticity
Synaptic vesicles are thought to be part of three distinct pools: the readily releasable pool (comprises approximately 5% of total vesicles), the recycling pool (about 15%), and the reserve pool (the remaining 80%).[10] The reserve pool seems to only begin to release vesicles in response to intense stimulation. There have been several studies that suggest the reserve vesicles are seldom ever released in response to physiological stimuli which raises questions about their importance.[10] This release in vesicles, regardless of which pool they are released from, is considered a form of short term synaptic plasticity because it is changing the functional characteristics of the presynaptic cell ultimately temporarily altering its firing properties. The difference between this and long-term potentiation is the fact that this phenomenon only occurs for the duration of time it takes to recycle and reuse neurotransmitters as opposed to it occurring over the long-term such as the characteristics underlying long-term potentiation. Further research should be conducted to identify the importance of the reserve pool vesicles in presynaptic cells.
Role in CNS pathologies
Synaptic Fatigue has not been shown to directly cause or result in a central nervous system pathology, although the degrees at which it is activated in cells has been studied as result of particular pathologies and diseases. Long-term changes in a neuron or synapse, resulting in a permanent change in a neuron's excitatory properties can cause synaptic fatigue to occur from much more or less activation that could potentially lead to some sort of physiological abnormality.
Alzheimer's Disease
Hallmarks of Alzheimer's disease (AD) are impairment of cognition, aggregation of β-amyloid peptide (Aβ), neurofibrillary degeneration, loss of neurons with accelerated atrophy of specific brain areas, and decrease of synapse number in surviving neurons. Research indicates both pre- and postsynaptic mechanisms resulting in AD. One specific abnormality includes an increased amount of presynaptic protein APP. A study was conducted where synaptic fatigue was compared between transgenic mice overexpressing APP/PS1 with their littermates who did not overexpress the protein. The results showed that fatigue was more significantly pronounced in the APP/PS1 mice, which indicates a decrease in the amount of readily releasable pools of vesicles in the presynaptic neuron. Conclusions from this study include synaptic fatigue being primarily a presynaptic phenomenon and not being affected by postsynaptic receptor desensitization, synaptic fatigue is not a result of Ca2+ ions building up in the terminal, and most importantly that synaptic fatigue is an important player and can be studied when researching the causes and effects of some neurodegenerative diseases.[3]
Depression
Antidepressants have short-term and long-term effects in depressed patients. The short-term effects are explained by a hypothesis that states that depression is acutely brought on by an immediate decrease in catecholamines in the brain. Antidepressants act immediately to inhibit this decrease and restore normal levels of these neurotransmitters in the brain. Under stressed conditions, vesicle exocytosis is potentiated and a release of catecholamines causes depression of presynaptic cells because of depleted neurotransmitters. Therapeutic doses of fluoxetine have been shown to decrease these neuronal fatigue states by inhibiting vesicle release and thereby preventing synaptic fatigue in hippocampal neurons. These findings show that fluoxetine as well as other antidepressants that act through the same mechanisms as fluoxetine enhance neurorecovery and neurotransmission to reduce the risk of depression.[11]
Unanswered questions
- Although now synaptic fatigue is thought to primarily be a presynaptic phenomenon, could postsynaptic processes account for a larger portion of the causes that are currently understood for synaptic fatigue?
- Recycling of synaptic-vesicle membrane proteins is rapid, as indicated by the ability of many neurons to fire fifty times a second, and quite specific, in that several membrane proteins unique to the synaptic vesicles are specifically internalized by endocytosis. Endocytosis usually involves clathrin-coated vesicles, though non-clathrin-coated vesicles may also be used. After the endocytic vesicles lose their clathrin coat, however, they usually do not fuse with larger, low pH endosomes, as they do during endocytosis of plasma-membrane proteins in other cells (see Figure 17-46). Rather, the recycled vesicles are immediately refilled with neurotransmitter.
http://www.ncbi.nlm.nih.gov/books/NBK21521/
References
- ↑ Kilpatrick, Z. P. (2010). Spatially Structured Waves and Oscillations in Neuronal Networks With Synaptic Depression and Adaptation. Doctor of Philosophy, University of Utah.
- 1 2 Simons-Weidenmaier, N. S., Weber, M., Plappert, C. F., Pilz, P. K. D., & Schmid, S. (2006). Synaptic depression and short-term habituation are located in the sensory part of the mammalian startle pathway. BMC Neuroscience, 7, 38-38.
- 1 2 Zhang, H., Gong, B., Liu, S., Fa, M., Ninan, I., Staniszewski, A., & Arancio, O. (2005). Synaptic fatigue is more pronounced in the APP/PS1 transgenic mouse model of Alzheimer's disease. Current Alzheimer Research, 2(2), 137-140.
- ↑ Hay, M., Hoang, C. J., & Pamidimukkala, J. (2001). Cellular mechanisms regulating synaptic vesicle exocytosis and endocytosis in aortic baroreceptor neurons. Annals Of The New York Academy Of Sciences, 940, 119-131.
- 1 2 Nadim, F., Manor, Y., Kopell, N., & Marder, E. (1999). Synaptic depression creates a switch that controls the frequency of an oscillatory circuit. Proceedings Of The National Academy Of Sciences Of The United States Of America, 96(14), 8206-8211.
- 1 2 Ryan, T. A., Smith, S. J., & Reuter, H. (1996). The timing of synaptic vesicle endocytosis. Proceedings Of The National Academy Of Sciences Of The United States Of America, 93(11), 5567-5571.
- ↑ Armbruster, M., & Ryan, T. A. (2011). Synaptic vesicle retrieval time is a cell-wide rather than individual-synapse property. [Article]. Nature Neuroscience, 14(7), 824-826. doi:10.1038/nn.2828
- ↑ Castellucci, V. F., & Kandel, E. R. (1974). A quantal analysis of the synaptic depression underlying habituation of the gill-withdrawal reflex in Aplysia. Proceedings Of The National Academy Of Sciences Of The United States Of America, 71(12), 5004-5008.
- 1 2 Abrahamsson, T., Gustafsson, B., & Hanse, E. (2005). Synaptic fatigue at the naive perforant path-dentate granule cell synapse in the rat. The Journal Of Physiology, 569(Pt 3), 737-750.
- 1 2 Ikeda, K., & Bekkers, J. M. (2009). Counting the number of releasable synaptic vesicles in a presynaptic terminal. [Article]. Proceedings Of The National Academy Of Sciences Of The United States Of America, 106(8), 2945-2950. doi:10.1073/pnas.0811017106
- ↑ Henkel, A. W., Welzel, O., Groemer, T. W., Tripal, P., Rotter, A., & Kornhuber, J. (2010). Fluoxetine prevents stimulation-dependent fatigue of synaptic vesicle exocytosis in hippocampal neurons. [Article]. Journal of Neurochemistry, 114(3), 697-705. doi:10.1111/j.1471-4159.2010.06795.x