Peptide synthesis
In organic chemistry, peptide synthesis is the production of peptides, which are organic compounds in which multiple amino acids are linked via amide bonds, also known as peptide bonds. The biological process of producing long peptides (proteins) is known as protein biosynthesis.
Chemistry
Peptides are synthesized by coupling the carboxyl group of one amino acid to the amino group of another amino acid molecule. Due to the possibility of unintended reactions, protecting groups are usually necessary. Chemical peptide synthesis most commonly starts at the carboxyl end of the peptide, and proceeds toward the amino-terminus. This is the opposite direction of protein biosynthesis.
Liquid-phase synthesis
Liquid-phase peptide synthesis is a classical approach to peptide synthesis. It has been replaced in most labs by solid-phase synthesis (see below). However, it retains usefulness in large-scale production of peptides for industrial purposes.
Solid-phase synthesis
Solid-phase peptide synthesis (SPPS), pioneered by Robert Bruce Merrifield,[1] caused a paradigm shift within the peptide synthesis community, and it is now the standard method for synthesizing peptides and proteins in the lab. SPPS allows for the synthesis of natural peptides which are difficult to express in bacteria, the incorporation of unnatural amino acids, peptide/protein backbone modification, and the synthesis of D-proteins, which consist of D-amino acids.
Small porous beads are treated with functional units ('linkers') on which peptide chains can be built. The peptide will remain covalently attached to the bead until cleaved from it by a reagent such as anhydrous hydrogen fluoride or trifluoroacetic acid. The peptide is thus 'immobilized' on the solid-phase and can be retained during a filtration process while liquid-phase reagents and by-products of synthesis are flushed away.
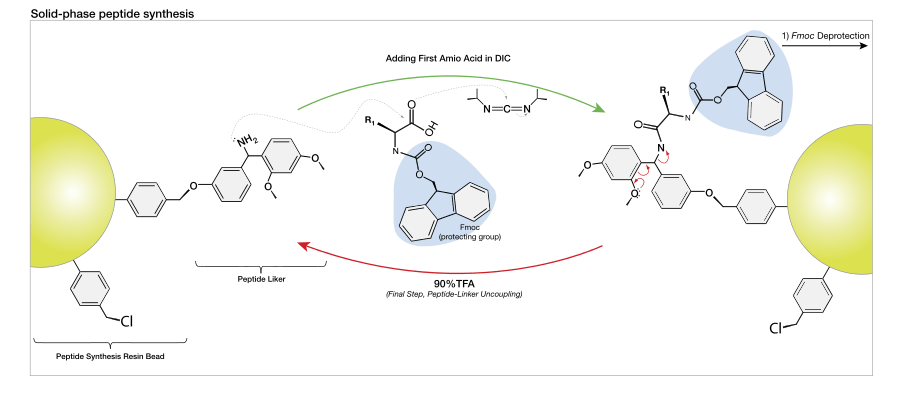
The general principle of SPPS is one of repeated cycles of deprotection-wash-coupling-wash. The free N-terminal amine of a solid-phase attached peptide is coupled (see below) to a single N-protected amino acid unit. This unit is then deprotected, revealing a new N-terminal amine to which a further amino acid may be attached. The superiority of this technique partially lies in the ability to perform wash cycles after each reaction, removing excess reagent with all of the growing peptide of interest remaining covalently attached to the insoluble resin.
The overwhelmingly important consideration is to generate extremely high yield in each step. For example, if each coupling step were to have 99% yield, a 26-amino acid peptide would be synthesized in 77% final yield (assuming 100% yield in each deprotection); if each step were 95%, it would be synthesized in 25% yield. Thus each amino acid is added in major excess (2~10x) and coupling amino acids together is highly optimized by a series of well-characterized agents.
There are two majorly used forms of SPPS – Fmoc and Boc. Unlike ribosome protein synthesis, solid-phase peptide synthesis proceeds in a C-terminal to N-terminal fashion. The N-termini of amino acid monomers is protected by either of these two groups and added onto a deprotected amino acid chain. Automated synthesizers are available for both techniques, though many research groups continue to perform SPPS manually.
SPPS is limited by yields, and typically peptides and proteins in the range of 70 amino acids are pushing the limits of synthetic accessibility. Synthetic difficulty also is sequence dependent; typically amyloid peptides and proteins are difficult to make. Longer lengths can be accessed by using native chemical ligation to couple two peptides together with quantitative yields.
Since its introduction over 40 years ago, SPPS has been significantly optimized. First, the resins themselves have been optimized.[2] Furthermore, the 'linkers' between the C-terminal amino acid and polystyrene resin have improved attachment and cleavage to the point of mostly quantitative yields.[3][4][5] The evolution of side chain protecting groups has limited the frequency of unwanted side reactions. In addition, the evolution of new activating groups on the carboxyl group of the incoming amino acid have improved coupling and decreased epimerization. Finally, the process itself has been optimized. In Merrifield's initial report, the deprotection of the α-amino group resulted in the formation of a peptide-resin salt, which required neutralization with base prior to coupling. The time between neutralization of the amino group and coupling of the next amino acid allowed for aggregation of peptides, primarily through the formation of secondary structures, and adversely affected coupling. The Kent group reported that concomitant neutralization of the α-amino group and coupling of the next amino acid led to improved coupling.[6] Each of these improvements has helped SPPS become the robust technique that it is today.
Solid supports
The name solid support implies that reactions are carried out on the surface of the support, but this is not the case. Reactions also occur within these particles, and thus the term "solid support" better describes the insolubility of the polymer. The physical properties of the solid support, and the applications to which it can be utilized, vary with the material from which the support is constructed, the amount of cross-linking, as well as the linker and handle being used. Most scientists in the field believe that supports should have the minimum amount of cross-linking to confer stability. This should result in a well-solvated system where solid-phase peptide synthesis can be carried out. Nonetheless, the characteristics of an efficient solid support include:[7] It must be physically stable and permit the rapid filtration of liquids, such as excess reagents, it must be inert to all reagents and solvents used during SPPS, it must swell extensively in the solvents used to allow for penetration of the reagents, and tt must allow for the attachment of the first amino acid.
There are three primary types of solid supports: gel-type supports, surface-type supports, and composites.[7] Gel-type supports are highly solvated polymers with an equal distribution of functional groups. This type of support is the most common, and includes polystyrene (styrene cross-linked with 1–2% divinylbenzene), polyacrylamide (hydrophilic alternative to polystyrene), polyethylene glycol (PEG) (PEG-polystyrene (PEG-PS) is more stable than polystyrene and spaces the site of synthesis from the polymer backbone), and PEG-based supports composed of a PEG-polypropylene glycol network or PEG with polyamide or polystyrene. Surface-type supports: Many materials have been developed for surface functionalization, including controlled pore glass, cellulose fibers, and highly cross-linked polystyrene. Composites are gel-type polymers supported by rigid matrices.
Polystyrene resin
.png)
Polystyrene resin is a versatile resin and it is quite useful in multi-well, automated peptide synthesis, due to its minimal swelling in dichloromethane. The initial support used by R. Bruce Merrifield was polysytrene cross-linked with 2% divinylbenzene. This support is sometimes referred to as the 'Merrifield resin.' This resin produces a hydrophobic bead that is solvated by a nonpolar solvent such as dichloromethane or dimethylformamide. Since then, new resins have been developed that have the advantages of chemical inertness, and enhanced swelling or rigidity (a property of mechanical strength). For instance, highly cross-linked (50%) polystyrene has been developed that possesses the features of increased mechanical stability, better filtration of reagents and solvents, and rapid reaction kinetics.
Polystyrene resins are also available as PEG hybrids. An example of this type of resin is the Tentagel resin. The base resin is polystyrene onto which is attached long chains (MW ca. 3000 Da) of polyethylene glycol (PEG; also known as polyethylene oxide). Synthesis is carried out on the distal end of the PEG spacer making it suited for long and difficult peptides. In addition, it is also attractive for the synthesis of combinatorial Peptide libraries and on resin screening experiments. It does not expand much during synthesis, making it a preferred resin for robotic peptide synthesis.
PEG-based resin
Improvements to solid supports used for peptide synthesis enhance their ability to withstand the repeated use of TFA during the deprotection step of SPPS.[8] Furthermore, different resins allow for different functional groups at the C-terminus. The oxymethylphenylacetamidomethyl (PAM) resin results in the conventional C-terminal carboxylic acid. On the other hand, the paramethylbenzhydrylamine (pMBHA) resin yields a C-terminal amide, which is useful in mimicking the interior of a protein.
Along with the development of Fmoc SPPS, different resins have also been created to be removed by TFA. Similar to the Boc strategy, two primary resins are used, based on whether a C-terminal carboxylic acid or amide is desired. The Wang resin was, as of 1996, the most commonly used resin for peptides with C-terminal carboxylic acids.[9] If a C-terminal amide is desired, the Rink amide resin is used.
Protecting groups
Amino acids have reactive α-carboxylic acid and α-amine groups that allow for their linking into polymers, but that also complicate the aim coupling specific pairs of amino acids, in precise order. In addition, many amino acids have reactive side chain functional groups, which can also react in a variety of ways, including with free α-carboxylic and α-amino groups during peptide synthesis (given the very reactive reagents present), "side-reactions" that would negatively influence yield and purity. Chemical groups have been developed to facilitate the synthesis of peptides with precise amino acid sequences, with minimal side reactions, groups that block or "protect" all functional groups present in amino acids except that pair whose coupling is desired. These protecting groups, while very many in practice, can be described in three groups: α-carboxylic acid (C-terminal) protecting groups, α-amino (N-terminal) protecting groups, and side chain protecting groups.
C-terminal protecting groups
Protecting groups of the carboxylic acid are the least used given the direction and methods applied in solid-supported peptide synthesis, but are covered first because to the brevity of the topic. These protecting groups are mostly used in liquid-phase synthesis, and there is some redundancy in the chemistry with respect to groups used to protect carboxylates in side chains (see below).
N-terminal protecting groups
Protected amino acids being added in a given step of peptide synthesis are added in excess to ensure maximal yields during each synthesis step. Without N-terminal protection of the added amino acid, its self-coupling (polymerization) would compete with the desired peptide synthesis, resulting in low yield of even failure of the desired peptide synthesis. N-terminal protection requires an additional step of removing the protecting group in each peptide synthesis cycle, a deprotection step, prior to the next coupling step.
As of this date, two protecting groups, tert-butyloxycarbonyl (t-Boc) and 9H-fluoren-9-ylmethoxycarbonyl (Fmoc) are most commonly used to protect the α-amine group of the "incoming" (newly added) amino acid in a solid-phase peptide synthesis cycle.
t-Boc and Fmoc protecting groups
Tert-butyloxycarbonyl (t-Boc) protection
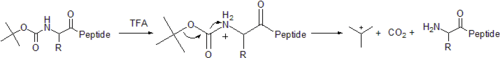
The original method for the synthesis of proteins relied on tert-butyloxycarbonyl (or more simply "Boc") to temporarily protect the α-amino group. In this method, the Boc group is covalently bound to the amino group to suppress its nucleophilicity. The C-terminal amino acid is covalently linked to the resin through a linker. Next, the Boc group is removed with acid, such as trifluoroacetic acid (TFA). This forms a positively charged amino group (in the presence of excess TFA; note image on the right illustrates neutral amino group), which is neutralized (via in-situ or non-in-situ methods) and coupled to the incoming activated amino acid.[6] Reactions are driven to completion by the use of excess (2- to 4-fold) activated amino acid. After each deprotection and coupling step, a wash with dimethylformamide (DMF) is performed to remove excess reagents, allowing (by year 2000) for high yields (~99%) during each cycle.[7]
t-Boc protecting strategies retain usefulness in reducing peptide aggregation during synthesis. t-Boc groups can be added to amino acids with t-Boc anhydride and a suitable base. Some researchers prefer Boc SPPS for complex syntheses . In addition, when synthesizing nonnatural peptide analogs, which are base-sensitive (such as depsipeptides), the t-Boc protecting group is necessary, because Fmoc SPPS uses a base to deprotect the α-amino group.
Permanent side-chain protecting groups are typically benzyl or benzyl-based groups. Final removal of the peptide from the linkage occurs simultaneously with side-chain deprotection with anhydrous hydrogen fluoride via hydrolytic cleavage. The final product is a fluoride salt which is relatively easy to solubilize. Importantly, scavengers such as cresol are added to the HF in order to prevent reactive t-butyl cations from generating undesired products. In fact, the use of harsh hydrogen fluoride may degrade some peptides, which was the premise for the development of a milder, base-labile method of SPPS—namely, the Fmoc method.
9H-fluoren-9-ylmethoxycarbonyl (Fmoc) protection
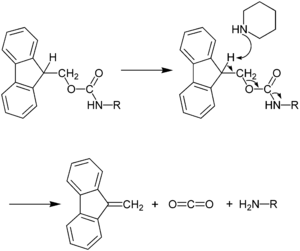
The capacity for anhydrous hydrogen fluoride to degrade proteins during the final cleavage conditions led to a new α-amino protecting group based on 9-fluorenylmethyloxycarbonyl (Fmoc). The Fmoc method allows for a milder deprotection scheme. This method utilizes a base, usually piperidine (20–50%) in DMF in order to remove the Fmoc group to expose the α-amino group for reaction with an incoming activated amino acid.[7] Unlike the acid used to deprotect the α-amino group in Boc methods, Fmoc SPPS uses a base, and thus the exposed amine is neutral. Therefore, no neutralization of the peptide-resin is required, but the lack of electrostatic repulsions between the peptides can lead to increased aggregation. Because the liberated fluorenyl group is a chromophore, deprotection by Fmoc can be monitored by UV absorbance of the runoff, a strategy which is employed in automated synthesizers.
The advantage of Fmoc is that it is cleaved under very mild basic conditions (e.g. piperidine), but stable under acidic conditions, although this has not always held true in certain synthetic sequences. This allows mild acid-labile protecting groups that are stable under basic conditions, such as Boc and benzyl groups, to be used on the side-chains of amino acid residues of the target peptide. This orthogonal protecting group strategy is common in organic synthesis. Fmoc is preferred over BOC due to ease of cleavage; however it is less atom-economical, as the fluorenyl group is much larger than the tert-butyl group. Accordingly, prices for Fmoc amino acids were high until the large-scale piloting of one of the first synthesized peptide drugs, enfuvirtide, began in the 1990s, when market demand adjusted the relative prices of the two sets of amino acids.
Semipermanent side chain protecting groups are t-butyl-based, and final cleavage of the protein from the resin and removal of permanent protecting groups is performed with TFA in the presence of scavengers. Water and triisopropylsilane (TIPS) present in a 1:1 ratio are often used as scavengers. Thus, the Fmoc method is orthogonal in two directions: deprotection of any α-amino group, deprotection of side groups and final cleavage from the resin occur by independent mechanisms. The resulting final product is a TFA salt, which is more difficult to solubilize than the fluoride salts generated in Boc SPPS. This method is thus milder than the Boc method because the deprotection/cleavage-from-resin steps occur with different conditions rather than with different reaction rates.
Comparison of t-Boc and Fmoc solid-phase peptide synthesis
Both the Fmoc and Boc methods offer advantages and disadvantages. The selection of one technique over another is thus made on a case-by-case basis.[11]
| Boc | Fmoc | |
|---|---|---|
| Requires special equipment | Yes | No |
| Cost of reagents | Lower | Higher |
| Solubility of peptides | Higher | Lower |
| Purity of hydrophobic peptides | High | May be lower |
| Problems with aggregation | Less frequently | More frequently |
| Synthesis time | ~20 min/amino acid | ~20–60 min/amino acid |
| Cleavage from resin | HF | TFA |
| Safety | Potentially dangerous | Relatively safe |
| Orthogonal | No | Yes |
Boc SPPS uses special equipment to handle the final cleavage and deprotection step, which requires anhydrous hydrogen fluoride. Because the final cleavage of the peptide with Fmoc SPPS uses TFA, this special equipment is not necessary. The solubility of peptides generated by Boc SPPS is generally higher than those generated with the Fmoc method, because fluoride salts are higher in solubility than TFA salts. Next, problems with aggregation are generally more of an issue with Fmoc SPPS, primarily because the removal of a Boc group with TFA yields a positively charged α-amino group, whereas the removal of an Fmoc group yields a neutral α-amino group. The steric hindrance of the positively charged α-amino group limits the formation of secondary structure on the resin. Finally, the Fmoc method is considered orthogonal, since α-amino group deprotection is with base, while final cleavage from the resin is with acid. The Boc method utilizes acid for both deprotection and cleavage from the resin. Hence, both methods possess advantages and disadvantages for their application in specific situations, and several factors must be considered to decide between the methods.
Other protecting groups
Benzyloxy-carbonyl
The (Z) group is another carbamate-type amine protecting group, first used by Max Bergmann in the synthesis of oligopeptides.[12] It is removed under harsh conditions using HBr in acetic acid, or milder conditions of catalytic hydrogenation. While it has been used periodically for α-amine protection in peptide synthesis, of this date, it is almost exclusively used for side chain protection.
Alloc and miscellaneous groups
The allyloxycarbonyl (alloc) protecting group is sometimes used to protect an amino group (or carboxylic acid or alcohol group) when an orthogonal deprotection scheme is required. It is also sometimes used when conducting on-resin cyclic peptide formation, where the peptide is linked to the resin by a side-chain functional group. The Alloc group can be removed using tetrakis(triphenylphosphine)palladium(0).
For special applications like synthetic steps involving protein microarrays, protecting groups sometimes termed "lithographic" are used, which are amenable to photochemistry at a particular wavelength of light, and so which can be removed during lithographic types of operations.
Side chain protecting groups
Amino acid side chains represent a broad range of functional groups and are sites of nonspecific reactivity during peptide synthesis. Because of this, many different protecting groups are required that are usually based on the benzyl (Bzl) or tert-butyl (tBu) group.[7] The specific protecting groups used during the synthesis of a given peptide vary depending on the peptide sequence and the type of N-terminal protection used (see next paragraph). Side chain protecting groups are known as permanent or semipermanent protecting groups, because they can withstand the multiple cycles of chemical treatment during synthesis and are only removed during treatment with strong acids after peptide synthesis is completed.
Because N-terminal deprotection occurs repeatedly during peptide synthesis, protecting schemes are established such that different types of side chain protecting groups (e.g., Bzl, tBu, etc.) are matched to either Boc or Fmoc, respectively, for ultimate deprotection that is optimized. Because multiple protecting groups are normally used during peptide synthesis of peptides greater in length than oligomers, care must be taken such that all side chain protecting groups are compatible, so that when deprotection of individual protecting groups is required (e.g., to selectively modify the side chain of one amino acid of a synthetic peptide), the one deprotection step does not affect other side chains. Protecting schemes are therefore developed in each particular case of a peptide synthesis to assign protecting groups to each amino acid residue.
Activating groups
For coupling the peptides the carboxyl group is usually activated. This is important for speeding up the reaction. There are two main types of activating groups: carbodiimides and triazolols. However the use of pentafluorophenyl esters (FDPP,[13] PFPOH[14]) and BOP-Cl[15] are useful for cyclising peptides.
Carbodiimides
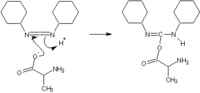
These activating agents were first developed. Most common are dicyclohexylcarbodiimide (DCC) and diisopropylcarbodiimide (DIC). Reaction with a carboxylic acid yields a highly reactive O-acylisourea. During artificial protein synthesis (such as Fmoc solid-state synthesizers), the C-terminus is often used as the attachment site on which the amino acid monomers are added. To enhance the electrophilicity of carboxylate group, the negatively charged oxygen must first be "activated" into a better leaving group. DCC is used for this purpose. The negatively charged oxygen will act as a nucleophile, attacking the central carbon in DCC. DCC is temporarily attached to the former carboxylate group (which is now an ester group), making nucleophilic attack by an amino group (on the attaching amino acid) to the former C-terminus (carbonyl group) more efficient. The problem with carbodiimides is that they are too reactive and that they can therefore cause racemization of the amino acid.
Triazoles
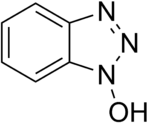
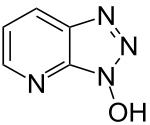
To solve the problem of racemization, triazoles were introduced. The most important ones are 1-hydroxy-benzotriazole (HOBt) and 1-hydroxy-7-aza-benzotriazole (HOAt), though others have also been developed. These substances can react with the O-acylurea to form an active ester which is less reactive and less in danger of racemization. HOAt is especially favourable because of a neighbouring group effect.[16] As of this date, HOBt has been removed from many chemical vendor catalogues; although almost always found as a hydrate, HOBt may be explosive when allowed to fully dehydrate and shipment by air or sea is heavily restricted. Alternatives to HOBt and HOAt have been introduced. One of the most promising and inexpensive is ethyl 2-cyano-2-(hydroxyimino)acetate (trade name Oxyma Pure), which is not explosive and has a reactivity of that in between HOBt and HOAt.
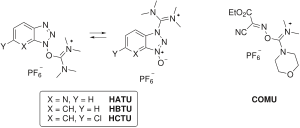
Newer developments omit the carbodiimides totally: the active ester is introduced as a uronium or phosphonium salt of a non-nucleophilic anion (tetrafluoroborate or hexafluorophosphate): HBTU, HATU, HCTU, TBTU, PyBOP. Two uronium types of the coupling additive of Oxyma Pure is also available as COMU or TOTU reagent.
Regioselective disulfide bond formation
The formation of multiple native disulfides remains one of the primary challenges of native peptide synthesis by solid-phase methods. Random chain combination typically results in several products with nonnative disulfide bonds.[17] Stepwise formation of disulfide bonds is typically the preferred method, and performed with thiol protecting groups.[18] Different thiol protecting groups provide multiple dimensions of orthogonal protection. These orthogonally protected cysteines are incorporated during the solid-phase synthesis of the peptide. Successive removal of these groups, to allow for selective exposure of free thiol groups, leads to disulfide formation in a stepwise manner. The order of removal of the groups must be considered so that only one group is removed at a time. Using this method, Kiso and coworkers reported the first total synthesis of insulin in 1993.[19]
Thiol protecting groups used in peptide synthesis requiring later regioselective disulfide bond formation must possess multiple characteristics. First, they must be reversible with conditions that do not affect the unprotected side chains. Second, the protecting group must be able to withstand the conditions of solid-phase synthesis. Third, the removal of the thiol protecting group must be such that it leaves intact other thiol protecting groups, if orthogonal protection is desired. That is, the removal of PG A should not affect PG B. Some of the thiol protecting groups commonly used include the acetamidomethyl (Acm), tert-butyl (But), 3-nitro-2-pyridine sulfenyl (NPYS), 2-pyridine-sulfenyl (Pyr), and triphenylmethyl (Trt) groups. Importantly, the NPYS group can replace the Acm PG to yield an activated thiol.[20]
Important to the discussion of disulfide bond formation is the order in which disulfides are formed. The synthesis insulin by Kiso and coworkers is illustrative of the logic and methods for regioselective disulfide bond formation. In this work, the A-chain of insulin was prepared with following protecting groups in place on its cysteines: CysA6(But), CysA7(Acm), and CysA11(But), leaving CysA20 unprotected.[19] Synthesis of the B-chain was performed with the following protecting groups in place CysB7(Acm) and CysB19(Pyr). The first disulfide bond, CysA20–CysB19, was formed by mixing the two chains in 8 M urea, pH 8 (RT) for 50 min, while the second disulfide bond, CysA7–CysB7, was formed by treatment with iodine in aqueous acetic acid to remove the Acm groups.[19] The third disulfide, the intramolecular CysA6–CysA11, was formed after the removal of the But groups by methyltrichlorosilane with diphenyl sulfoxide in TFA.[19] Importantly, formation of the first disulfide in 8 M urea, pH 8 does not affect the other protecting groups, namely Acm and But groups. Likewise, formation of the second disulfide bond with iodine in aqueous acetic acid does not affect the But groups. From a logical standpoint, the order in which the thiol groups are exposed to form disulfides should be of little consequence, since the other cysteines are protected; however, it is observed, practically, that the order in which disulfides are formed can have a significant effect on yields.[21]
Synthesizing long peptides
Stepwise elongation, in which the amino acids are connected step-by-step in turn, is ideal for small peptides containing between 2 and 100 amino acid residues. Another method is fragment condensation, in which peptide fragments are coupled. Although the former can elongate the peptide chain without racemization, the yield drops if only it is used in the creation of long or highly polar peptides. Fragment condensation is better than stepwise elongation for synthesizing sophisticated long peptides, but its use must be restricted in order to protect against racemization. Fragment condensation is also undesirable since the coupled fragment must be in gross excess, which may be a limitation depending on the length of the fragment.
A new development for producing longer peptide chains is chemical ligation: unprotected peptide chains react chemoselectively in aqueous solution. A first kinetically controlled product rearranges to form the amide bond. The most common form of native chemical ligation uses a peptide thioester that reacts with a terminal cysteine residue. Other methods applicable for covalently linking polypeptides in aqueous solution include the use of split inteins,[22] spontaneous isopeptide bond formation[23] and sortase ligation.[24]
In order to optimize synthesis of long peptides, Zealand Pharma (located in Denmark in Medicon Valley) invented a method for converting a difficult peptide sequence into an easy peptide sequence. The new technology, called SIP-technology, uses “structure-inducing probes” (SIP) to facilitate the synthesis of long peptides. The SIP-technology is a small pre-sequence peptide sequence (e.g. Lysine (Lysn); Glutamic Acid (Glun); (LysGlu)n) that is incorporated at the C-terminus of subsequent resin bound peptide to induce an alpha-helix-like structure in the peptide. The SIP technology constrains the parent peptide into a more ordered conformation using intramolecular hydrogen bonds. This allows the peptide structure to stabilize, and the utilized hydrogen bonds reduce the likelihood of aggregation and degradation by enzymes. In this way, the SIP technology is designed to optimize peptide synthesis, increase biological half-life, improve peptide stability and inhibit enzymatic degradation without altering pharmacological activity or profile of action.[25][26]
Microwave-assisted peptide synthesis
Although microwave irradiation has been around since the late 1940s, it was not until 1986 that microwave energy was used in organic chemistry. During the end of the 1980s and 1990s, microwave energy was an obvious source for completing chemical reactions in minutes that would otherwise take several hours to days. Through several technical improvements at the end of the 1990s and beginning of the 2000s, microwave synthesizers have been designed to provide both low and high energy pockets of microwave energy so that the temperature of the reaction mixture could be controlled. The microwave energy used in peptide synthesis is of a single frequency providing maximum penetration depth of the sample which is in contrast to conventional kitchen microwaves.
In peptide synthesis, microwave irradiation has been used to complete long peptide sequences with high degrees of yield and low degrees of racemization. Microwave irradiation during the coupling of amino acids to a growing polypeptide chain is catalyzed not only by the increase in temperature but also by the alternating electric field of the microwave.[27] This is because the polar N-terminal amine group and peptide backbone continuously try to align with the alternating electric field, thus helping prevent aggregation and increasing access to the solid phase reaction matrix. This increases yields of the final peptide products. There is however no clear evidence that microwave is better than simple heating and some peptide laboratories regard microwave just as a convenient method for rapid heating of the peptidyl resin. Heating to above 50–55 degrees Celsius also prevents aggregation and accelerates the coupling.
Despite the main advantages of microwave irradiation of peptide synthesis, the main disadvantage is the racemization which may occur with the coupling of cysteine and histidine. A typical coupling reaction with these amino acids are performed at lower temperatures than the other 18 natural amino acids. A number of peptides do not survive microwave synthesis or heating in general. One of the more serious side effects is dehydration (loss of water) which for certain peptides can be almost quantitative like pancreatic polypeptide (PP). This side effect is also seen by simple heating without the use of microwave.
Cyclic peptides
On resin cyclization
Peptides can be cyclized on a solid support. A variety of cylization reagents can be used such as HBTU/HOBt/DIEA, PyBop/DIEA, PyClock/DIEA. Head-to-tail peptides can be made on the solid support. The deprotection of the C-terminus at some suitable point allows on-resin cyclization by amide bond formation with the deprotected N-terminus. Once cyclization has taken place, the peptide is cleaved from resin by acidolysis and purified. The strategy for the solid-phase synthesis of cyclic peptides in not limited to attachment through Asp, Glu or Lys side chains. Cysteine has a very reactive sulfhydryl group on its side chain. A disulfide bridge is created when a sulfur atom from one Cysteine forms a single covalent bond with another sulfur atom from a second cysteine in a different part of the protein. These bridges help to stabilize proteins, especially those secreted from cells. Some researchers use modified cysteines using S-acetomidomethyl (Acm) to block the formation of the disulfide bond but preserve the cysteine and the protein's original primary structure.
Off-resin cyclization
Off-resin cyclization is a solid-phase synthesis of key intermediates, followed by the key cyclization in solution phase, the final deprotection of any masked side chains is also carried out in solution phase. This has the disadvantages that the efficiencies of solid-phase synthesis are lost in the solution phase steps, that purification from by-products, reagents and unconverted material is required, and that undesired oligomers can be formed if macrocycle formation is involved.[28]
See also
References
- ↑ R. B. Merrifield (1963). "Solid Phase Peptide Synthesis. I. The Synthesis of a Tetrapeptide". J. Am. Chem. Soc. 85 (14): 2149–2154. doi:10.1021/ja00897a025.
- ↑ Mitchell, A. R. K., S.B.H.; Engelhard, M.; Merrifield, R.B. (1978). "A new synthetic route to tert-butyloxycarbonylaminoacyl-4-(oxymethyl)phenylacetamidomethyl-resin, an improved support for solid-phase peptide synthesis". J. Org. Chem. 43 (13): 2845–2852. doi:10.1021/jo00408a022.
- ↑ Wang, S.-S. (1973). "p-alkoxybenzyl alcohol resin and p-alkoxybenzyloxycarbonylhydrazide resin for solid phase synthesis of protected peptide fragments.". J. Am. Chem. Soc. 95 (4): 1328–33. doi:10.1021/ja00785a052. PMID 4687686.
- ↑ Matsueda, G. R. a. S., J.M. (1981). "A p-methylbenzylhydrlamine resin for improved solid-phase synthesis of peptide amides". Peptides. 2 (1): 45–50. doi:10.1016/S0196-9781(81)80010-1. PMID 7243625.
- ↑ Sieber, P. (1987). "A new acid-labile anchor group for the solid-phase synthesis of C-terminal peptide amides by the Fmoc method". Tetrahedron Lett. 28: 2107–10. doi:10.1016/S0040-4039(00)96055-6.
- 1 2 Schnolzer, M. A., P.; Jones, A.; Alewood, D.; Kent, S.B.H. (2007). "In Situ Neutralization in Boc-chemistry Solid Phase Peptide Synthesis". Int. J. Peptide Res. Therap. 13 (1–2): 31–44. doi:10.1007/s10989-006-9059-7.
- 1 2 3 4 5 Albericio, F. (2000). Solid-Phase Synthesis: A Practical Guide (1 ed.). Boca Raton: CRC Press. p. 848. ISBN 0824703596.
- ↑ Feinberg, R. S.; Merrifield, R. B. (1974). "Zinc chloride-catalyzed chloromethylation of resins for solid phase peptide synthesis". Tetrahedron. 30 (17): 3209–3212. doi:10.1016/S0040-4020(01)97575-1.
- ↑ Hermkens, P. H. H.; Ottenheijm, H. C. J.; Rees, D. C. (1997). "Solid-phase organic reactions II: A review of the literature Nov 95 – Nov 96". Tetrahedron. 53 (16): 5643–5678. doi:10.1016/S0040-4020(97)00279-2.
- ↑ Jones, J. (1992). Amino Acid and Peptide Synthesis. Oxford, UK: Oxford University Press.
- ↑ Nilsson BL, Soellner MB, Raines RT (2005). "Chemical Synthesis of Proteins". Annu. Rev. Biophys. Biomol. Struct. 34: 91–118. doi:10.1146/annurev.biophys.34.040204.144700. PMC 2845543
 . PMID 15869385.
. PMID 15869385. - ↑ Bergmann, Max & Zervas, Leonidas (1932). "Über ein allgemeines Verfahren der Peptid-Synthese". Berichte der deutschen chemischen Gesellschaft. 65 (7): 1192–1201. doi:10.1002/cber.19320650722.
- ↑ K. C. Nicolaou; Natarajan, Swaminathan; Li, Hui; Jain, Nareshkumar F.; Hughes, Robert; Solomon, Michael E.; Ramanjulu, Joshi M.; Boddy, Christopher N. C.; Takayanagi, Masaru (1998). "Total Synthesis of Vancomycin Aglycon – Part 1: Synthesis of Amino Acids 4–7 and Construction of the AB-COD Ring Skeleton". Angew. Chem. Int. Ed. 37 (19): 2708–2714. doi:10.1002/(SICI)1521-3773(19981016)37:19<2708::AID-ANIE2708>3.0.CO;2-E.
- ↑ Schmidt; Joullié, Madeleine M. (1998). "Synthetic studies of 14-membered cyclopeptide alkaloids". Tetrahedron Lett. 39 (40): 7211–7214. doi:10.1016/S0040-4039(98)01589-5.
- ↑ R. Baker; J. L. Castro (1989). "The total synthesis of (+)-macbecin I". Chem. Commun. (6): 378–381. doi:10.1039/C39890000378.
- ↑ L. A. Carpino (1993). "1-Hydroxy-7-azabenzotriazole. An efficient peptide coupling additive". J. Am. Chem. Soc. 115 (10): 4397–4398. doi:10.1021/ja00063a082.
- ↑ Zhang, J.-W.; Wu, Cui Rong; Liu, Wen; Zhang, Jing Wen (1991). "Disulfide bond formation in peptides by dimethyl sulfoxide. Scope and applications". J. Am. Chem. Soc. 113 (17): 6657–6662. doi:10.1021/ja00017a044.
- ↑ Sieber, P.; Kamber, B.; Hartmann, A.; Jöhl, A.; Riniker, B.; Rittel, W. (1977). "Total synthesis of human insulin. IV. Description of the final steps (author's transl)". Helvetica Chimica Acta. 60 (1): 27–37. doi:10.1002/hlca.19770600105. PMID 838597.
- 1 2 3 4 Akaji, K.; Fujino, K.; Tatsumi, T.; Kiso, Y. (1993). "Total synthesis of human insulin by regioselective disulfide formation using the silyl chloride-sulfoxide method". Journal of the American Chemical Society. 115 (24): 11384–11392. doi:10.1021/ja00077a043.
- ↑ Ottl, J.; Battistuta, R.; Pieper, M.; Tschesche, H.; Bode, W.; Kuhn, K.; Moroder, L. (1996). "Design and synthesis of heterotrimeric collagen peptides with a built-in cystine-knot. Models for collagen catabolism by matrix-metalloproteases". FEBS Lett. 398 (1): 31–36. doi:10.1016/S0014-5793(96)01212-4. PMID 8946948.
- ↑ This may be because the formation of the CysA20–CysB19 disulfide may place the thiol group of CysB7 in close proximity with both CysA6 and CysA7, leading to multiple disulfide products.
- ↑ Aranko AS, Wlodawer A, Iwaï H (2014). "Nature's recipe for splitting inteins". Prot Eng Des Sel. 27: 263–71. doi:10.1093/protein/gzu028. PMC 4133565. PMID 25096198.
- ↑ Reddington SC, Howarth M (2015). "Secrets of a covalent interaction for biomaterials and biotechnology: SpyTag and SpyCatcher". Curr Op Chem Biol. 29: 94–9. doi:10.1016/j.cbpa.2015.10.002. PMID 26517567.
- ↑ Haridas V, Sadanandan S, Dheepthi NU (2014). "Sortase-based bio-organic strategies for macromolecular synthesis". Chembiochem. 15: 1857–67. doi:10.1002/cbic.201402013. PMID 25111709.
- ↑ Kapusta, D. R.; Thorkildsen, C; Kenigs, VA; Meier, E; Vinge, MM; Quist, C; Petersen, JS (2005). "Pharmacodynamic Characterization of ZP120 (Ac-RYYRWKKKKKKK-NH2), a Novel, Functionally Selective Nociceptin/Orphanin FQ Peptide Receptor Partial Agonist with Sodium-Potassium-Sparing Aquaretic Activity". Journal of Pharmacology and Experimental Therapeutics. 314 (2): 652–60. doi:10.1124/jpet.105.083436. PMID 15855355.
- ↑ Rizzi A, Rizzi D, Marzola G, et al. (October 2002). "Pharmacological characterization of the novel nociceptin/orphanin FQ receptor ligand, ZP120: in vitro and in vivo studies in mice". Br. J. Pharmacol. 137 (3): 369–74. doi:10.1038/sj.bjp.0704894. PMC 1573505. PMID 12237257.
- ↑ Palasek, Stacey A.; Cox, Zachary J.; and Collins, Jonathan M. (2007). "Limiting racemization and aspartimide formation in microwave-enhanced Fmoc solid phase peptide synthesis". Journal of Peptide Science. 13 (3): 143–148. doi:10.1002/psc.804. PMID 17121420.
- ↑ Peter Scott (13 October 2009). Linker Strategies in Solid-Phase Organic Synthesis. John Wiley & Sons. pp. 135–137. ISBN 978-0-470-74905-0.
Further reading
- Stewart, J.M.; Young, J.D. (1984). Solid phase peptide synthesis (2nd ed.). Rockford, IL: Pierce Chemical Company. p. 91. ISBN 0935940030.
- Kent, Stephen B. H. (1984). "Chemical Synthesis of Peptides and Proteins". Annual Review of Biochemistry. 57. Palo Alto, CA: Annual Reviews. pp. 957–989. doi:10.1146/annurev.bi.57.070188.004521. Retrieved 12 November 2016.
- Atherton, E.; Sheppard, R.C. (1989). Solid Phase peptide synthesis: a practical approach. Oxford, England: IRL Press. ISBN 0199630674.
- Chan, W. & White, Peter, eds. (2000). Fmoc Solid Phase Peptide Synthesis: A Practical Approach. Practical Approach Series, Issue 222. Oxford, UK: Oxford University Press. ISBN 0199637245. Retrieved 12 November 2016.
- Stawikowski, Maciej & Fields, Gregg B. (2002). "Unit–18.1. Introduction to Peptide Synthesis". Curr Protoc Protein Sci. doi:10.1002/0471140864.ps1801s26. PMC 3564544. Retrieved 12 November 2016.
- Bodanszky, M. (2012). Principles of Peptide Synthesis. Reactivity and Structure: Concepts in Organic Chemistry, Volume 16. New York, NY: Springer Science & Business Media. ISBN 3642967639. Retrieved 12 November 2016.
- Bodanszky, M. & Bodanszky, A. (2013). The Practice of Peptide Synthesis. Reactivity and Structure: Concepts in Organic Chemistry, Volume 21. New York, NY: Springer Science & Business Media. ISBN 364296835X. Retrieved 12 November 2016.
- Benoiton, N. Leo (2016). Chemistry of Peptide Synthesis. Boca Raton, FL: CRC Press / Taylor & Frances. ISBN 1420027697. Retrieved 12 November 2016.
- Laconde, Guillaume & Desroses, Matthieu (2016). "Synthetic Protocols Coupling Reagents in Amide Synthesis" (commercial blog). Montpellier, France: Helixem. ISBN 1420027697. Retrieved 12 November 2016.
External links
| Wikimedia Commons has media related to Peptide synthesis. |