Phosphine oxide
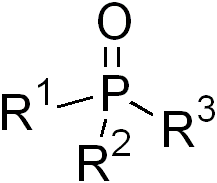
Phosphine oxides are phosphorus compounds with the formula OPX3. When X = alkyl or aryl, these are organophosphine oxides. Triphenylphosphine oxide is an example. An inorganic phosphine oxide is phosphoryl chloride (POCl3). Such compounds are thermally stable, decomposing only above 450 °C.[1] Phosphoryl refers to a functional group drawn with a phosphorus-oxygen double bond.
Structure and bonding
Phosphine oxides feature tetrahedral phosphorus centers. The P-O bond is short and polar. According to Molecular Orbital Theory, the short P-O bond is attributed to the donation of the lone pair electrons from oxygen p-orbitals to the antibonding phosphorus-carbon bonds; This proposal, which is supported by ab initio calculations, has gained consensus in the chemistry community.[2]
The nature of the P-O bond was once hotly debated. Some discussions invoked a role for phosphorus-centered d-orbitals in bonding, but this analysis is not supported by computational analyses. In terms of simple Lewis structure, the bond is more accurately represented as a dative bond, as is currently used to depict an amine oxide.
Syntheses
Phosphine oxides are generated as a by-product of the Wittig reaction:
- R3PCR'2 + R"2CO → R3PO + R'2C=CR"2
Another route to phosphine oxides is the thermolysis of phosphonium hydroxides:
- [PPh4]Cl + NaOH → Ph3PO + NaCl + PhH
In the laboratory, phosphine oxides are usually generated by the oxidation, often accidentally, of tertiary phosphines:
- R3P + 1/2 O2 → R3PO
Basic phosphines, such as trialkyl derivatives, are prone to this reaction.
The hydrolysis of phosphorus(V) dihalides also affords the oxide:[3]
- R3PCl2 + H2O → R3PO + 2 HCl
A special nonoxidative route is applicable secondary phosphine oxides, which arise by the hydrolysis of the chlorophosphine. An example is the hydrolysis of chlorodiphenylphosphine to give diphenylphosphine oxide:
- Ph2PCl + H2O → Ph2P(O)H + HCl
Deoxygenation
One common way of synthesizing tertiary phosphine ligands is by deoxygenation of phosphine oxides (OPR3) with trichlorosilane. Deoxygenation proceeds with retention or inversion of configuration, depending on conditions. Inversion is favored by a combination of trichlorosilane and triethylamine. In the absence of the base, the reaction proceeds with retention. The stereochemical course is explained mechanistically by complexation followed by intramolecular hydride transfer. On the other hand, the inversion of configuration in the presence of triethylamine is explained by complexation followed by intermolecular hydride transfer from a 1:1 triethylamine-trichlorosilane complex in an SN2 reaction. Based on these observations, it is thought that complexation of OPR3 by trichlorosilane occurs with retention of configuration.[4]
- HSiCl3 + Et3N ⇋ −SiCl3 + Et3N+H
- R3PO + Et3NH+ ⇋ R3P+OH + Et3N
- −SiCl3 + R3P + OH → PR3 + HOSiCl3
The popularity of this method is partly attributable to the availability of inexpensive trichlorosilane. Instead of HSiCl3, other perchloropolysilanes, e.g. hexachlorodisilane (Si2Cl6), can also be used to deoxygenate phosphine oxides. In comparison, using the reaction of the corresponding phosphine oxides with perchloropolysilanes such as Si2Cl6 or Si3Cl8 in benzene or chloroform, phosphines can be prepared in higher yields.
- R3PO + Si2Cl6 → R3P + Si2OCl6
- 2 R3PO + Si3Cl8 → 2 R3P + Si3O2Cl8
Deoxygenation with perchloropolysilanes or trichlorosilane requires longer reaction times and is often problematic with sterically hindered or electron-deficient phosphine oxides. To counter this problem Ph3P or (EtO)3P along with trichlorosilane can be used.[5]
Use
Phosphine oxides are ligands in various applications of homogeneous catalysis. In coordination chemistry, they are known to have labilizing effects to CO ligands cis to it in organometallic reactions. The cis effect describes this process.
Phosphine oxides are typically (and appropriately) viewed as useless by-products in the Wittig reaction. They can, however, be usable in certain Wittig-like reactions. Thus, benzaldehyde has been converted to β-methoxystyrene using methoxymethyl diphenylphosphine oxide in a two step procedure. In step one the phosphine oxide is deprotonated at −90 °C in THF/ether with lithium diisopropylamide, then the aldehyde is added. After aqueous workup, adducts are isolated. With potassium-t-butoxide the adducts are, at room temperature, converted to the styrenes. As the adducts exist as a separatable mixture of D/L compounds and a meso-form the final styrenes are obtainable as pure E- or Z-form .[6]
Parent compound
The parent compound phosphine oxide (H3PO) is unstable. It has been detected with mass spectrometry as a reaction product of oxygen and phosphine,[7] by means of FT-IR in a phosphine-ozone reaction [8] and in matrix isolation with a reaction of phosphine, vanadium oxytrichloride and chromyl chloride.[9] It has also been reported relatively stable in a water-ethanol solution by electrochemical oxidation of white phosphorus, where it slowly disproportionates into phosphine and hypophosphorous acid.[10] Phosphine oxide is tautomeric with phosphinous acid (H2POH).

Phosphine oxide is reported as an intermediate in the room-temperature polymerization of phosphine and nitric oxide to solid PxHy.[11]
References
- ↑ D. E. C. Corbridge "Phosphorus: An Outline of its Chemistry, Biochemistry, and Technology" 5th Edition Elsevier: Amsterdam 1995. ISBN 0-444-89307-5.
- ↑ D. B. Chesnut (1999). "The Electron Localization Function (ELF) Description of the PO Bond in Phosphine Oxide". Journal of the American Chemical Society. 121 (10): 2335–2336. doi:10.1021/ja984314m.
- ↑ W. B. McCormack (1973). "3-Methyl-1-Phenylphospholene oxide". Org. Synth.; Coll. Vol., 5, p. 787
- ↑ Klaus Naumann; Gerald Zon; Kurt Mislow (1969). "Use of hexachlorodisilane as a reducing agent. Stereospecific deoxygenation of acyclic phosphine oxides". Journal of the American Chemical Society. 91: 7012–7023. doi:10.1021/ja01053a021.
- ↑ Hai-Chen Wu; Jin-Quan Yu; Jonathan B. Spencer (2004). "Stereospecific Deoxygenation of Phosphine Oxides with Retention of Configuration Using Triphenylphosphine or Triethyl Phosphite as an Oxygen Acceptor". Organic Letters. 6: 4675–4678. doi:10.1021/ol048227c.
- ↑ Synthesis of 3- (1 -alkenyloxy) -1,2-propanediols, enol ethers of glycerol, by the horner-wittig reaction Recueil des Travaux Chimiques des Pays-Bas Volume 102, Issue 10, 1983, Pages: 465–466, T. A. M. van Schaik and A. van der Gen doi:10.1002/recl.19831021009
- ↑ Kinetics and mechanism of the reactions of PH3 with O(3P) and N(4S) atoms Peter A. Hamilton and Timothy P. Murrells J. Chem. Soc., Faraday Trans. 2, 1985, 81, 1531-1541 doi:10.1039/F29858101531
- ↑ FTIR spectra of the photolysis products of the phosphine-ozone complex in solid argon Robert. Withnall, Lester. Andrews J. Phys. Chem., 1987, 91 (4), pp 784–797 doi:10.1021/j100288a008
- ↑ Matrix Isolation and Theoretical Study of the Photochemical Reaction of PH3 with OVCl3 and CrCl2O2 David A. Kayser and Bruce S. Ault J. Phys. Chem. A, 2003, 107 (33), pp 6500–6505 doi:10.1021/jp022692e
- ↑ Yakhvarov, D.; Caporali, M.; Gonsalvi, L.; Latypov, S.; Mirabello, V.; Rizvanov, I.; Sinyashin, O.; Stoppioni, P.; Peruzzini, M. (2011). "Experimental Evidence of Phosphine Oxide Generation in Solution and Trapping by Ruthenium Complexes". Angewandte Chemie International Edition. 50: 5370–5373. doi:10.1002/anie.201100822.
- ↑ Phosphine Polymerization by Nitric Oxide: Experimental Characterization and Theoretical Predictions of Mechanism Yi-Lei Zhao, Jason W. Flora, William David Thweatt, Stephen L. Garrison, Carlos Gonzalez, K. N. Houk and Manuel Marquez Inorg. Chem., 2009, 48 (3), pp 1223–1231 doi:10.1021/ic801917a