Organocopper compound

Organocopper compounds in organometallic chemistry contain carbon to copper chemical bonds. Organocopper chemistry is the science of organocopper compounds describing their physical properties, synthesis and reactions.[1][2][3] They are reagents in organic chemistry.
The first organocopper compound, the explosive copper(I) acetylide Cu2C2 (Cu-C≡C-Cu), was synthesized by Rudolf Christian Böttger in 1859 by passing acetylene gas through copper(I) chloride solution:[4]
- C2H2 + 2 CuCl → Cu2C2 + 2 HCl
Structure and bonding
Organocopper compounds are diverse in structure and reactivity, but organocopper compounds are largely limited in oxidation states to copper(I), sometimes denoted Cu+1. As a d10 metal center, it is related to Ni(0), but owing to its higher oxidation state, it engages in less pi-backbonding. Organic derivatives of Cu(II) and Cu(III) are invoked as intermediates but rarely isolated or even observed. In terms of geometry, copper(I) adopts symmetrical structures, in keeping with its spherical electronic shell. Typically one of three coordination geometries is adopted: linear 2-coordinate, trigonal 3-coordinate, and tetrahedral 4-coordinate. Organocopper compounds form complexes with a variety of soft ligands such as alkylphosphines (R3P), thioethers (R2S), and cyanide (CN−).
Simple complexes with CO, alkene, and Cp ligands
Copper(I) salts have long been known to bind CO, albeit weakly. A representative complex is CuCl(CO), which is polymeric. In contrast to classical metal carbonyls, pi-backbonding is not strong in these compounds.[5]

Alkenes bind to copper(I), although again generally weakly. The binding of ethylene to Cu in proteins is of broad significance in plant biology so much so that ethylene is classified as a plant hormone. Its presence, detected by the Cu-protein, affects ripening and many other developments.[6]
Although copper does not form a metallocene, half-sandwich complexes can be produced. One such derivative is (η-cyclopentadienyl triethylphosphine) copper.[7] This compound is a relatively rare example of an organocopper complex that follows the 18-electron rule.
Alkyl and aryl copper compounds
Copper halides react with organolithium reagents to give organocopper compounds. The area was pioneered by Henry Gilman, who reported methylcopper in 1936. Thus, phenylcopper is prepared by reaction of phenyllithium with copper(I) bromide in diethyl ether. Grignard reagents can be used in place of organolithium compounds. Gilman also investigated the dialkylcuprates. These are obtained by combining two equivalent of RLi with Cu(I) salts. Alternatively, these cuprates are prepared from oligomeric neutral organocopper compounds by treatment with one equivalent of organolithium reagent.
Compounds of the type [CuRn](n-1)- are reactive towards oxygen and water, forming copper(I) oxide. They also tend to be thermally unstable, which can be useful in certain coupling reactions. Despite these difficulties, organocopper reagents are frequently generated and consumed in situ with no attempt to isolate them. They are used very frequently in organic chemistry as alkylating reagents because they exhibit greater functional group tolerance than corresponding Grignard and organolithium reagents. The electronegativity of copper is much higher than its next-door neighbor in the group 12 elements, zinc, suggesting diminished nucleophilicity for its carbon ligands.
Copper salts react with terminal alkynes to form the acetylides.
Structures of simple alkyl and aryl copper compounds
Alkyl and aryl copper complexes aggregate both in crystalline form and in solution. Aggregation is especially evident for charge-neutral organocopper compounds, i.e. species with the empirical formula (RCu), which adopt cyclic structures. Since each copper center requires at least two ligands, the organic group is a bridging ligand. This effect is illustrated by the structure of mesitylpentacopper, which is a pentamer. A cyclic structure is also seen for CuCH2SiMe3, first 1:1 organocopper compound to be analyzed by X-ray crystallography (1972 by Lappert). This compound is relatively stable because the bulky trimethylsilyl groups provide steric protection. It is a tetramer, forming an 8-membered ring with alternating Cu-C bonds. In addition the four copper atoms form a planar Cu4 ring based on three-center two-electron bonds. The copper to copper bond length is 242 pm compared to 256 pm in bulk copper. In pentamesitylpentacopper a 5-membered copper ring is formed, similar to (2,4,6-Trimethylphenyl)gold, and pentafluorophenylcopper is a tetramer.[8]

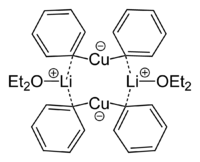
Lithium dimethylcuprate is a dimer in diethyl ether forming an 8-membered ring with two lithium atoms linking two methyl groups. Similarly, lithium diphenylcuprate forms a dimeric etherate, [{Li(OEt2)}(CuPh2)]2, in the solid state.[9]
Reactions of organocuprates
Cross-coupling reactions
Prior to the development of palladium-catalyzed cross coupling reaction, copper was the preferred catalyst for almost a century. Palladium offers faster more selective reaction. Although in the recent years copper compounds have appeared again as a synthetically useful as well as an eco-friendly metal, its low cost.[10]
Reactions of R2CuLi with alkyl halides R'-X gives the coupling product:
- R2CuLi + R'X → R-R' + CuR + LiX
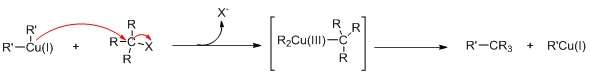
The reaction mechanism involve oxidative addition (OA) of the alkyl halide to Cu(I) elevating it to give a planar Cu(III) intermediate, followed by reductive elimination (RE). The nucleophilic attack is the rate-determining step. In the case for substitution of iodide, single electron transfer mechanism is proposed (see figure).
![{\displaystyle [{\ce {R}}{-}{\color {Blue}{\ce {Cu}}}{\ce {-R}}]^{-}{\ce {Li+}}\ {\xrightarrow {\color {Red}{\ce {R'-X}}}}\ \left[{\ce {R}}{-}{\overset {{\displaystyle \color {Red}{\ce {R}}'} \atop |}{\underset {| \atop {\displaystyle \color {Red}{\ce {X}}}}{\color {Blue}{\ce {Cu}}}}}{\ce {-R}}\right]^{-}{\ce {Li+}}{\ce {->R}}{-}{\color {Blue}{\ce {Cu}}}+{\ce {R}}{-}{\color {Red}{\ce {R'}}}+{\ce {Li}}{-}{\color {Red}{\ce {X}}}}](../I/m/9d7db6776d965a97de00837677a94c6fd3d1df2a.svg)
Many electrophiles participate in this reaction. The approximate order of reactivity, beginning with the most reactive, is as follows: acid chlorides[11] aldehydes > tosylates ~ epoxides > iodides > bromides > chlorides > ketones > esters > nitriles >> alkenes
Generally the OA-RE mechanism is analogous to palladium-catalyzed cross coupling reactions. One difference between copper and palladium is that copper can undergo single electron transfer processes.[12]

Coupling reactions
Oxidative coupling is the coupling of copper acetylides to conjugated alkynes in the Glaser coupling (for example in the synthesis of cyclooctadecanonaene) or to aryl halides in the Castro-Stephens Coupling.
Reductive coupling is a coupling reaction of aryl halides with a stoichiometric equivalent of copper metal that occurs in the Ullmann reaction. In an example of a present-day cross coupling reaction called decarboxylative coupling, a catalytic amount of Cu(I) displaces a carboxyl group forming the arylcopper (ArCu) intermediate. Simultaneously, a palladium catalyst converts an aryl bromide to the organopalladium intermediate (Ar'PdBr), and on transmetallation the biaryl is formed from ArPdAr'.[13][14]

Redox neutral coupling is the coupling of terminal alkynes with halo-alkynes with a copper(I) salt in the Cadiot-Chodkiewicz coupling. Thermal coupling of two organocopper compounds is also possible.
Conjugate addition
In 1941, Kharash discovered that Grignard reagents add to cyclohexenone in presence of Cu(I) resulting in 1,4-addition instead of 1,2-addition.[15] This work foreshadowed extensive studies on the conjugate additions to enones with organocuprates. Note that if a Grignard reagent (such as RMgBr) is used, the reaction with an enone would instead proceed through a 1,2-addition.[16] The 1,4-addition mechanism of cuprates to enones goes through the nucleophilic addition of the Cu(I) species at the beta-carbon of the alkene to form a Cu(III) intermediate, followed by reductive elimination of Cu(I).[17] In the original paper describing this reaction, methylmagnesium bromide is reacted with isophorone with and without 1 mole percent of added copper(I) chloride (see figure).[15]

Without added salt the main products are alcohol B (42%) from nucleophilic addition to the carbonyl group and diene C (48%) as its dehydration reaction product. With added salt the main product is 1,4-adduct A (82%) with some C (7%).
A 1,6-addition is also possible, for example in one step of the commercial-scale production of fulvestrant:[18]

Cu(III) intermediates
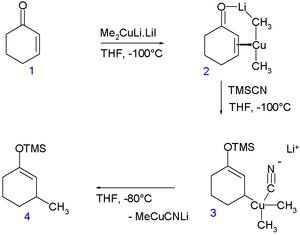
The involvement of the otherwise rare Cu(III) oxidation state has been demonstrated in the conjugate addition of the Gilman reagent to an enone:[19] In a so-called rapid-injection NMR experiment at -100 °C, the Gilman reagent Me2CuLi (stabilized by lithium iodide) was introduced to cyclohexenone (1) enabling the detection of the copper — alkene pi complex 2. On subsequent addition of trimethylsilyl cyanide the Cu(III) species 3 is formed (indefinitely stable at that temperature) and on increasing the temperature to -80 °C the conjugate addition product 4. According to an accompanying in silico experiments [20] the Cu(III) intermediate has a square planar molecular geometry with the cyano group in cis orientation with respect to the cyclohexenyl methine group and anti-parallel to the methine proton. With other ligands than the cyano group this study predicts room temperature stable Cu(III) compounds.
Carbocupration
Carbocupration is a nucleophilic addition of organocopper reagents (R-Cu) to acetylene or terminal alkynes resulting in an alkenylcopper compound (RC=C-Cu).[21] It is a special case of carbometalation and also called the Normant reaction.[22]
Figure: Catalytic cycle for carbocupration Muller,.[23]

Synthetic applications
Ullman chemistry
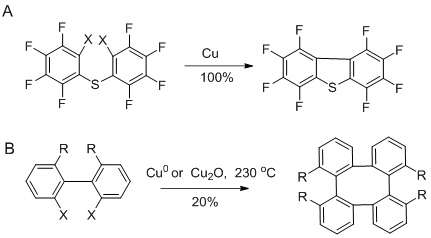
Ullman used Goldeberg design synthesis to develop copper based reactions that enabled the formation of C-C, C-N and C-S bonds.[24] Ullmann chemistry is based on the formation of a carbon-carbon bond via condensation of aryl halides in the presence of a copper compound. This type of reaction has been useful for ring closures, aryl bond formation, synthesis of symmetrical and unsymmetrical biaryl compounds, synthesis of oligophenylenes and so on.
There are two types of Ullman reaction: Classic (Copper catalyzed synthesis of symmetric biaryl compounds) and Ullman type (copper catalyzed nucleophilic aromatic substitution). Electronegative groups in the ortho position of the aryl halogen are known to be strongly activated toward Ullman reaction.[25] On the other hand, this reaction is inhibited by the steric hindrance provided by bulky groups on ortho positions of the aryl groups.
Ullman condensation has been used to afford linear polyphenilene compounds as shown in the image below.

Ullman synthesis of biaryl compounds can be employed to asymmetric reaction conditions.[26] Nelson and collaborators worked on the synthesis of asymmetric biaryl compounds and obtained the thermodynamically controlled product.[26]

The diastereometric rate of the product is enhanced with bulkier R groups in the auxiliary oxazoline group. Tert-butyl group has higher level of selectivity in the Ullman Coupling. Using an oxazoline auxiliary group provides steric effect which influences the high diastereoselectivity of asymmetric Ullman reaction.
Biphenyls had been obtained before with reasonable yields using 2, 2 diiodobiphenyl or 2, 2 diiodobiphenylonium ion as starting material; the reaction proceed when heating with Cu0 or Cu2O.

5-membered ring closures are reported to be favorable more facile, but larger rings have also been made.
Thermal dimerization proceeds via a copper hydride mechanism with complete retention of organocopper configuration.[12]

Oxidative dimerization proceeds via oxidation of dialkylcuprate I to neutral transient diakyl copper (II) which decomposes to give the desired akyl alkyl dimer. This reaction is first order kinetics for both the organocopper and the substrate with inversion as the stereochemical consequence.SN2 like mechanism for oxidative dimerization and direct displacement
Direct displacement
Reactions with alkyl halides and organocopper compound are examples of direct displacement mechanism. As in an SN2 reaction, inversion of configuration occurs, as in oxidative dimerization. On the other hand, reactions of organocopper compound with alkenyl halides proceed with retention of subtrate’s configuration. Two possibe mechanisms have been considered.[12] In the case of organocopper coupling with aryl halides the reaction proceed via aromatic nucleophilic substitution reaction. These reactions are reported to occur with lithium diaryl cuprates with a transmetalation step that forms mixed homocuprate compounds. The composition of this mixture can be generally determined statistically from the amount of substrate that is present before oxidation.
- Organocopper and aryl halides coupling general mechanism

Cross coupling and transmetallation reactions
Sonogashira cross coupling reaction
The Sonogashira cross coupling reaction utilizes copper as a [co-catalyst], and palladium as the main catalyst. Its main synthetically application is toward the coupling of aryl and/or vinyl halides with terminal alkynes. The Sonogashira reaction has provided efficient routes to synthesize cyclic alkynes, which are used for a variety of applications including click chemistry.[27]
'Synthesis of 2-amino-1-(2-propynyl)pyridinium bromide'.[27] One of many applications of the Sonogashira cross coupling reactions is in the synthesis of imidazopyridine derivatives which have a diverse range of biological activities.
Synthesis of imidazopyridine derivatives

A general mechanism for the Pd-Cu transmetallation on the Sonogashira cross coupling reaction is shown below. Sonogashira reactions have been modify to use other catalysts and co-catalyst metals, e.g. Palladium free Sonogashira reactions have been published using copper as the main catalyst,[28] although it is well known that palladium impurities can accelerates Sonogashira reaction rates.[29]
Proposed Sonogashira copper-palladium synergistic coupling of acetylene and aryl halides

It is unknown which reaction cycle proceeds first in this cross coupling mechanism, therefore we are uncertain on which of the cycle produce the coupled product. Sonogashira Pd-free reactions had already been developed; these reactions are economically favorable since the expensive metal is removed. Although it was demonstrated that this cross coupling reactions is very sensitive to Pd and even ppb can make a huge difference in the reaction rate. Pd act as an engine for the Sonogashira reaction while copper acts as fuel by coordination to acetylide.[29]
Palladium free, copper catalyzed Sonogashira reaction: proposed mechanism [28]

The use of CuI and a carboxamide ligand readily improves the efficiency of copper catalyze (Pd-Free) reaction, and it inhibited the cyclization by- products and thus provides regioselectivity.
Chan Lam coupling reaction
This reaction enables the formation of aryl carbon- hetoroatom bonds. The Chan Lam reaction is an oxidative coupling of boronic acids, stannanes or siloxanes with NH or OH containing compounds, the reaction is catalyzed by stoichiometric amounts of copper (II). It has been use as a milder approach, compared to Baeyer-Villiger oxidation, to afford direct esterification of carboxylic acids.[30] The reaction's stereochemistry is controlled by electronic by electronic substituents at the aryl group; electron donating groups on the aryl enhance the yield of the reaction.
Cu(OTf)2-mediated Chan-Lam reaction of carboxylic acids
RCOOH + ArB(OH)2 -> RCOOAr
Chan Lam reaction has also been useful for selective transformations e.g. s- arylation [31]
Copper based reduction reactions
Phosphine copper hydrides Copper hydrides are generally considered mild reducing agent which meaning that they can afford better selectivity in reaction.[23] Copper hydrides are used generally in organic synthesis as mild reducing agents. Their use became a hot area of study after Stryker reagent report on 1988 where he described the use of his reagents to selectively reduce the β position on α,β - unsaturated carbonyl derivatives.[23]
Stryker reagent The copper hydride compound [(PPH3)CuH]6 is known as the Stryker reagent. The Stryker reagent is used in organic reactions as a source of hydride ions and is generally used in conjugate addition reactions. Stryker’s reagent provides remarkable regioselectivity favoring the formation of 1, 4- addition products when reacted with α,β- unsaturated carbonyl compounds. .[32]

Buchwald copper reaction The Buchwald reaction is a copper-catalyzed asymmetric reduction of activated alkenes using bidentate ligands such as (S)-T- BINAP.[23][33]
Catalytic conjugate reduction of α,β unsaturated esters using Buchwald reagent

Other copper mediated reductions
Synthesis of Z- fluorolefins
More Specific applications of copper based catalyst leads to the stereoselective synthesis of Z-fluorolefin derivatives. Synthesis of Z-Fluoro alkene dipeptide isosteres,.[34][35] Other effort to make this a more selective reactions includes the use of oxidation reduction condition for the reaction.[36] Fluoride acts as a leaving group and it enhances regioselectivity in the transformation the Z- Fluoroalkene.

Cu alkylation reaction
γ- Alkylation of allylic alcohols.[37]
Generally, the alkylation reaction of organocopper reagents proceed via gamma- alkylation. Cis- gamma attack occurs better in cyclohexyl carbamate due to sterics.

Alkylation of amines using the Gilman reagent Yamamoto and coworkers described an efficient synthetic method for the alkylation of amines. The reaction is based on the oxidative coupling of lithium alkyl copper amide which is reported to form in situ during the reaction between lithium dialkylcuprates and primary or secondary amides.[38]
Amine alkylation reaction

The reaction is reported to be favorable in ethereal solvents. This method was proved to be very effective for the oxidative coupling of amines and alkyl, including tertbutyl, and aryl halides.[38]
Vicinal functionalization reactions
Vicinal functionalization using a Carbocupration- Mukaiyama aldol reaction sequence [39]
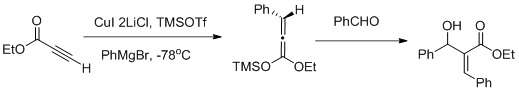
Muller and collaborators reported a vicinal functionalization of α,β- acetylenic esters using a Carbocupration/ Mukaiyama aldol reaction sequence (as shown in fig above) carbocupration favors the formation of the Z- aldol.
See also
- Chemistries of carbon with other elements of the periodic table:
| CH | He | ||||||||||||||||
| CLi | CBe | CB | CC | CN | CO | CF | Ne | ||||||||||
| CNa | CMg | CAl | CSi | CP | CS | CCl | CAr | ||||||||||
| CK | CCa | CSc | CTi | CV | CCr | CMn | CFe | CCo | CNi | CCu | CZn | CGa | CGe | CAs | CSe | CBr | CKr |
| CRb | CSr | CY | CZr | CNb | CMo | CTc | CRu | CRh | CPd | CAg | CCd | CIn | CSn | CSb | CTe | CI | CXe |
| CCs | CBa | CHf | CTa | CW | CRe | COs | CIr | CPt | CAu | CHg | CTl | CPb | CBi | CPo | CAt | Rn | |
| Fr | CRa | Rf | Db | CSg | Bh | Hs | Mt | Ds | Rg | Cn | Nh | Fl | Mc | Lv | Ts | Og | |
| ↓ | |||||||||||||||||
| CLa | CCe | CPr | CNd | CPm | CSm | CEu | CGd | CTb | CDy | CHo | CEr | CTm | CYb | CLu | |||
| Ac | CTh | CPa | CU | CNp | CPu | CAm | CCm | CBk | CCf | CEs | Fm | Md | No | Lr | |||
| Core organic chemistry | Many uses in chemistry |
| Academic research, but no widespread use | Bond unknown |
References
- ↑ Gary H. Posner (1980). An introduction to synthesis using organocopper reagents. New York: Wiley: Wiley. ISBN 0-471-69538-6.
- ↑ W.A. Herrmann, ed. (1999). Synthetic Methods of Organometallic and Inorganic Chemistry. 5, Copper, Silver, Gold, Zinc, Cadmium, and Mercury. Stuttgart: Thieme. ISBN 3-13-103061-5.
- ↑ Christoph Elschenbroich (2006). Organometallics (3 ed.). Weinheim: Wiley-VCH. ISBN 3-527-29390-6.
- ↑ R. C. Böttger (1859). "Ueber die Einwirkung des Leuchtgases auf verschiedene Salzsolutionen, insbesondere auf eine ammoniakalische Kupferchlorürlösung". Annalen. 109 (3): 351. doi:10.1002/jlac.18591090318.
- ↑ Strauss, S. H., "Copper(I) and Silver(I) Carbonyls. To be or not to be Nonclassical", Journal of the Chemical Society, Dalton Transactions, 2000, 1-6. doi:10.1039/A908459B
- ↑ Light, K. M.; Wisniewski, J. A.; Vinyard, W. A.; Kieber-Emmons, M. T., "Perception of the plant hormone ethylene: known-knowns and known-unknowns", J. Biol. Inorg. Chem. 2016, volume 21, 715-728. doi:10.1007/s00775-016-1378-3
- ↑ Delbaere, L. T. J.; McBride, D. W.; Ferguson, R. B. "Crystal structure of π-cyclopentadienyl(triethylphosphine)copper(I), π-C5H5CuP(C2H5)3" Acta Crystallographica, Section B 1970, volume 26 (Pt. 5), 515-21.
- ↑ Cairncross, Allan; Sheppard, William A; Wonchoba, Edward; Guilford, William J; House, Cynthia B; Coates, Robert M (1979). "Pentafluorophenylcopper tetramer, a reagent for synthesis of fluorinated aromatic compounds.". Organic Syntheses. 59: 122. doi:10.15227/orgsyn.059.0122.
- ↑ N. P. Lorenzen, E. Weiss (1990). "Synthesis and Structure of a Dimeric Lithium Diphenylcuprate:[{Li(OEt)2}(CuPh2)]2". Angew. Chem. Int. Ed. 29 (3): 300–302. doi:10.1002/anie.199003001.
- ↑ Yao, Bo.; Liu, Y.; Zhao, L.; Wang, D.; Wang, M. Designing a Cu(II)−ArCu(II)−ArCu(III)−Cu(I) Catalytic Cycle: Cu(II)-Catalyzed Oxidative Arene C−H Bond Azidation with Air as an Oxidant under Ambient Conditions. J. Org. Chem. 2014. Doi: doi.org/10.1021/jo502115a
- ↑ For an example see: Posner, Gary H.; Whitten, Charles E. (2003). "Secondary and Tertiary Alkyl Ketones from Carboxylic Acid Chlorides and Lithium Phenylthio(Alkyl)Cuprate Reagents:tert-Butyl Phenyl Ketone": 122–122. doi:10.1002/0471264180.os055.28.
- 1 2 3 Posner, G. H. 2011. Substitution Reactions Using Organocopper Reagents. Organic Reactions. 22:2:253–400
- ↑ Goossen, L. J.; Deng, G; Levy, LM (2006). "Synthesis of Biaryls via Catalytic Decarboxylative Coupling". Science. 313 (5787): 662–4. Bibcode:2006Sci...313..662G. doi:10.1126/science.1128684. PMID 16888137.
- ↑ Reagents: base potassium carbonate, solvent NMP, catalysts palladium acetylacetonate, Copper(I) iodide, MS stands for molecular sieves, ligand phenanthroline
- 1 2 3 Kharasch, M. S.; Tawney, P. O. (1941). "Factors Determining the Course and Mechanisms of Grignard Reactions. II. The Effect of Metallic Compounds on the Reaction between Isophorone and Methylmagnesium Bromide". Journal of the American Chemical Society. 63 (9): 2308–2316. doi:10.1021/ja01854a005. ISSN 0002-7863.
- ↑ For an example: Organic Syntheses, Coll. Vol. 9, p.328 (1998); Vol. 72, p.135 (1995) Link.
- ↑ Nakamura, Eiichi; Mori, Seiji (2000). "Wherefore Art Thou Copper? Structures and Reaction Mechanisms of Organocuprate Clusters in Organic Chemistry". Angewandte Chemie. 39 (21): 3750–3771. doi:10.1002/1521-3773(20001103)39:21<3750::AID-ANIE3750>3.0.CO;2-L. PMID 11091452.
- ↑ Fulvestrant: From the Laboratory to Commercial-Scale Manufacture Eve J. Brazier, Philip J. Hogan, Chiu W. Leung, Anne O’Kearney-McMullan, Alison K. Norton, Lyn Powell, Graham E. Robinson, and Emyr G. Williams Organic Process Research & Development 2010, 14, 544–552 doi:10.1021/op900315j
- 1 2 Bertz, Steven H.; Cope, Stephen; Murphy, Michael; Ogle, Craig A.; Taylor, Brad J. (2007). "Rapid Injection NMR in Mechanistic Organocopper Chemistry. Preparation of the Elusive Copper(III) Intermediate1". Journal of the American Chemical Society. 129 (23): 7208–9. doi:10.1021/ja067533d. PMID 17506552.
- ↑ Hu, Haipeng; Snyder, James P. (2007). "Organocuprate Conjugate Addition: The Square-Planar "CuIII" Intermediate". Journal of the American Chemical Society. 129 (23): 7210–1. doi:10.1021/ja0675346. PMID 17506553.
- ↑ For an example: Organic Syntheses, Coll. Vol. 7, p.236 (1990); Vol. 64, p.1 (1986) Link
- ↑ Normant, J; Bourgain, M. (1971). "Synthese stereospecifique and reactivite d' organocuivreux vinyliques". Tetrahedron Letters. 12 (27): 2583. doi:10.1016/S0040-4039(01)96925-4.
- 1 2 3 4 Cox, N.; Dang, H.; Whittaker, A.M.; Lalic, G. (2014). "NHC- copper hydrides as chemoselective reducing agents: catalytic reduction of alkynes, alkyl triflates and alkyl halides". Tetrahedron. 70: 4219–4231. doi:10.1016/j.tet.2014.04.004.
- ↑ Fanta, P.E. (1974). "The Ullmann Synthesis of Biaryls". Synthesis. 1974: 9–21. doi:10.1055/s-1974-23219.
- ↑ Beletkaya, I.P.; Cheprakov, A.V. (2004). "Copper in Cross Coupling Reactions: The Post Ullman Chemistry". Coord. Chem. Rev. 248: 2337–2364.
- 1 2 Nelson, T.D.; Meyers, A.I. (1994). "The asymmetric Ullman reaction, 2. The synthesis of enantiomerically pure C2-Symmetric Binaphtyls". J. Org. chem. 59: 2655–2658. doi:10.1021/jo00088a066.
- 1 2 Bakherad, M.; Nasr-Isfahani, H.; Keivanloo, A.; Doostmohammadi, N. Pd-Cu catalyzed heterocyclization during Sonogashira coupling: Synthesis of 2 benzylimidazo [1,2-α]pyridine. Tetrahedron Lett. 2008, 49, 3819-3822.
- 1 2 Jiang, H.; Fu, H.; Jiang, Y.; Zhao, Y. "Palladium free copper catalyzed Sonogashira Cross Coupling at room temperature". Synthesis. 2008: 2417–2426. doi:10.1055/s-2008-1067127.
- 1 2 Gonda, Z.; Tolnai, G.L.; Novák, Z. (2010). "Dramatic Impact of ppb levels of Palladium on the Copper catalyzed Sonogashira coupling". Chem. Eur J. 16: 11822–11826. doi:10.1002/chem.201001880.
- ↑ Zhang, L.; Zhang, G.; Zhang, M.; Cheng, J. (2010). "Cu(OTf)2-mediated Chan-Lam reaction of carboxylicacids to access phenolic esters". J. Org. Chem. 75: 7572–7474.
- ↑ Suvajit, K.; Chowdhury, S.; Chanda, T.; Ramul, B.J.; Anand, N.; Singh, S. (2014). "Ligand and base free Cu(II)- mediated selective S-Arylation of α-enolic dithioesters by Chan lam at room temperature". Eur J. Org. Chem. 0000: 0–0.
- ↑ Lipshutz, B.H.; Keith, J.; Papa, P.; Vivian, R. (1998). "A convenient, efficient method for conjugate reductions using catalytic quantities of CuI". Tetrahedron Lett. 39: 4627–4630. doi:10.1016/s0040-4039(98)00855-7.
- ↑ Jurkauskas, V.; Sadighi, J.P. Buchwald, S.L. Conjugate addition of a,b- unsaturated compounds catalyzad by a copper carbene complex. Org. lett. 2003, 5(14), 2417-2420
- ↑ Otaka, A.; Watanabe, H.; Mitsoyama, E.; Yukimasa, A.; Tamamura, H.; Fujii, N. Synthesis of (Z)-fluoroalkene isosteres utilizing organocopper- mediated reduction of gama, gamma- α,β - enoates. Tetrahedron Lett. 2001, 42, 285-287.
- ↑ Okada, M.; Nakamura, Y. Sago, A.; Hirokawa, H.; Taguchi, T. Stereoselective cosnstruction of functionalized (Z)- fluoroalkenes directed to o- depsipeptide isosteres. Tetrahedron Lett. 2003, 43, 5845-5847.
- ↑ Otaka, A.; Watanabe, H.; Yukimasa, A.; Oishi, S.; Tamamura, H.; Fuji, N. New access to α- substituted (Z)-fluoroalkene dipeptide isosteres utilizing organocopper reagents under redoctive-oxidative alkylation (R-OA) conditions. Tetrahedron Lett. 2001, 42, 5443-5446
- ↑ Yamamoto, Y.; Yamammoto, S.; Yatagai, H.; Maruyama, K (1980). "Lewis acid mediated reactions of organocopper reagent. A remarkably enhanced regioselective gamma- attack of allylic halides and direct alkylation of allylic alcohols via RCu.BF3". JACS. 102 (7): 2318–2325. doi:10.1021/ja00527a032.
- 1 2 Yamamoto, H.; Marouka, K. Novel N-alkylation of amines with organocopper reagents. J. Org. chem. 1980, 45, 2739-2740.
- ↑ Muller, A.J.; Jennings, M.P. Vicinal Functionalization of propionilate Esters via Tandem Catalytic Carbocupration-Mukaiyama Aldol Reaction sequence. Org. Lett. 2008, 10, 1649-1652