Organocatalysis
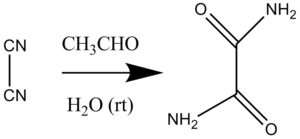
In organic chemistry, the term organocatalysis (a portmanteau of the terms "organic" and "catalyst") refers to a form of catalysis, whereby the rate of a chemical reaction is increased by an organic catalyst referred to as an "organocatalyst" consisting of carbon, hydrogen, sulfur and other nonmetal elements found in organic compounds.[3][4][5][6][7][8] Because of their similarity in composition and description, they are often mistaken as a misnomer for enzymes due to their comparable effects on reaction rates and forms of catalysis involved.
Organocatalysts which display secondary amine functionality can be described as performing either enamine catalysis (by forming catalytic quantities of an active enamine nucleophile) or iminium catalysis (by forming catalytic quantities of an activated iminium electrophile). This mechanism is typical for covalent organocatalysis. Covalent binding of substrate normally requires high catalyst loading (for proline-catalysis typically 20-30 mol%). Noncovalent interactions such as hydrogen-bonding facilitates low catalyst loadings (down to 0.001 mol%).
Organocatalysis offers several advantages. There is no need for metal-based catalysis thus making a contribution to green chemistry. In this context, simple organic acids have been used as catalyst for the modification of cellulose in water on multi-ton scale.[9] When the organocatalyst is chiral an avenue is opened to asymmetric catalysis, for example the use of proline in aldol reactions is an example of chirality and green chemistry.[10]
Introduction
Regular achiral organocatalysts are based on nitrogen such as piperidine used in the Knoevenagel condensation.[11] DMAP used in esterfications and DABCO used in the Baylis-Hillman reaction. Thiazolium salts are employed in the Stetter reaction. These catalysts and reactions have a long history but current interest in organocatalysis is focused on asymmetric catalysis with chiral catalysts, called asymmetric organocatalysis or enantioselective organocatalysis. A pioneering reaction developed in the 1970s is called the Hajos–Parrish–Eder–Sauer–Wiechert reaction. Between 1968 and 1997, there were only a few reports of the use of small organic molecules as catalysts for asymmetric reactions (the Hajos–Parrish reaction probably being the most famous), but these chemical studies were viewed more as unique chemical reactions than as integral parts of a larger, interconnected field.[12]
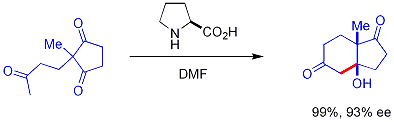
In this reaction, naturally occurring chiral proline is the chiral catalyst in an Aldol reaction. The starting material is an achiral triketone and it requires just 3% of proline to obtain the reaction product, a ketol in 93% enantiomeric excess. This is the first example of an amino acid-catalyzed asymmetric aldol reaction.[13][14]
The asymmetric synthesis of the Wieland-Miescher ketone (1985) is also based on proline and another early application was one of the transformations in the total synthesis of Erythromycin by Robert B. Woodward (1981).[15] A mini-review digest article focuses on selected recent examples of total synthesis of natural and pharmaceutical products using organocatalytic reactions.[16]
Many chiral organocatalysts are an adaptation of chiral ligands (which together with a metal center also catalyze asymmetric reactions) and both concepts overlap to some degree.
Organocatalyst classes
Organocatalysts for asymmetric synthesis can be grouped in several classes:
- Biomolecules: proline, phenylalanine. Secondary amines in general.[17] The cinchona alkaloids, certain oligopeptides.
- Synthetic catalysts derived from biomolecules.
- Hydrogen bonding catalysts, including TADDOLS, derivatives of BINOL such as NOBIN, and organocatalysts based on thioureas
- Triazolium salts as next-generation Stetter reaction catalysts
Examples of asymmetric reactions involving organocatalysts are:
- Asymmetric Diels-Alder reactions
- Asymmetric Michael reactions
- Asymmetric Mannich reactions
- Shi epoxidation
- Organocatalytic transfer hydrogenation
Proline
Proline catalysis has been reviewed.[18][19]
Imidazolidinone organocatalysis

A certain class of imidazolidinone compounds (also called MacMillan organocatalysts) are suitable catalysts for many asymmetric reactions such as asymmetric Diels-Alder reactions. The original such compound was derived from the biomolecule phenylalanine in two chemical steps (amidation with methylamine followed by condensation reaction with acetone) which leave the chirality intact:[20]

This catalyst works by forming an iminium ion with carbonyl groups of α,β-unsaturated aldehydes (enals) and enones in a rapid chemical equilibrium. This iminium activation is similar to activation of carbonyl groups by a Lewis acid and both catalysts lower the substrate's LUMO:[21]

The transient iminium intermediate is chiral which is transferred to the reaction product via chiral induction. The catalysts have been used in Diels-Alder reactions, Michael additions, Friedel-Crafts alkylations, transfer hydrogenations and epoxidations.
One example is the asymmetric synthesis of the drug warfarin (in equilibrium with the hemiketal) in a Michael addition of 4-hydroxycoumarin and benzylideneacetone:[22]

A recent exploit is the vinyl alkylation of crotonaldehyde with an organotrifluoroborate salt:[23]

For other examples of its use: see organocatalytic transfer hydrogenation and asymmetric Diels-Alder reactions.
Thiourea organocatalysis
A large group of organocatalysts incorporate the urea or the thiourea moiety. These catalytically effective (thio)urea derivatives termed (thio)urea organocatalysts provide explicit double hydrogen-bonding interactions to coordinate and activate H-bond accepting substrates.
References
- ↑ Justus von Liebig, Justus (1860). "Ueber die Bildung des Oxamids aus Cyan". Annalen der Chemie und Pharmacie. 113 (2): 246–247. doi:10.1002/jlac.18601130213.
- ↑ W. Langenbeck (1929). "Über organische Katalysatoren. III. Die Bildung von Oxamid aus Dicyan bei Gegenwart von Aldehyden". Liebigs Ann. 469: 16. doi:10.1002/jlac.19294690103.
- ↑ Berkessel, A., Groeger, H. (2005). Asymmetric Organocatalysis. Weinheim: Wiley-VCH. ISBN 3-527-30517-3.
- ↑ Special Issue: List, Benjamin (2007). "Organocatalysis". Chem. Rev. 107 (12): 5413–5883. doi:10.1021/cr078412e.
- ↑ Peter I. Dalko; Lionel Moisan (2004). "In the Golden Age of Organocatalysis". Angew. Chem. Int. Ed. 43 (39): 5138–5175. doi:10.1002/anie.200400650.
- ↑ Matthew J. Gaunt; Carin C.C. Johansson; Andy McNally; Ngoc T. Vo (2007). "Enantioselective organocatalysis". Drug Discovery Today. 12 (1/2): 8–27. doi:10.1016/j.drudis.2006.11.004. PMID 17198969.
- ↑ Dieter Enders; Christoph Grondal; Matthias R. M. Hüttl (2007). "Asymmetric Organocatalytic Domino Reactions". Angew. Chem. Int. Ed. 46 (10): 1570–1581. doi:10.1002/anie.200603129.
- ↑ Peter I. Dalko; Lionel Moisan (2001). "Enantioselective Organocatalysis". Angew. Chem. Int. Ed. 40 (20): 3726–3748. doi:10.1002/1521-3773(20011015)40:20<3726::AID-ANIE3726>3.0.CO;2-D.
- ↑ International Patent WO 2006068611 A1 20060629 " Direct Homogeneous and Heterogeneous Organic Acid and Amino Acid-Catalyzed Modification of Amines and Alcohols" Inventors: Armando Córdova, Stockholm, Sweden; Jonas Hafrén, Stockholm, Sweden.
- ↑ Example 4 in U.S. Patent 3,975,440 August 17, 1976, Filed Dec. 9, 1970 Zoltan G. Hajos and David R. Parrish.
- ↑ List, B. (2010). "Emil Knoevenagel and the Roots of Aminocatalysis". Angewandte Chemie International Edition in English. 49 (10): 1730–1734. doi:10.1002/anie.200906900. PMID 20175175.
- ↑ "The advent and development of organocatalysis" David C. MacMillan, NATUREVol 455|18 September 2008|doi:10.1038/nature07367
- ↑ Z. G. Hajos, D. R. Parrish, German Patent DE 2102623 1971
- ↑ Zoltan G. Hajos; David R. Parrish (1974). "Asymmetric synthesis of bicyclic intermediates of natural product chemistry". J. Org. Chem. 39 (12): 1615–1621. doi:10.1021/jo00925a003.
- ↑ R. B. Woodward; E. Logusch; K. P. Nambiar; K. Sakan; D. E. Ward; B. W. Au-Yeung; P. Balaram; L. J. Browne; et al. (1981). "Asymmetric total synthesis of erythromcin. 1. Synthesis of an erythronolide A secoacid derivative via asymmetric induction". J. Am. Chem. Soc. 103 (11): 3210–3213. doi:10.1021/ja00401a049.
- ↑ B. -F. Sun (2015). "Total synthesis of natural and pharmaceutical products powered by organocatalytic reactions". Tetrahedron Lett. 56 (17): 2133–2140. doi:10.1016/j.tetlet.2015.03.046.
- ↑ Organocatalysis—after the gold rush Søren Bertelsen and Karl Anker Jørgensen Chem. Soc. Rev., 2009, 38, 2178–2189 doi:10.1039/b903816g
- ↑ Gaunt, M. J.; Johansson, C. C. C.; McNally, A.; Vo, N. T. (2007). "Enantioselective organocatalysis". Drug Discovery Today. 12 (1–2): 8–27. doi:10.1016/j.drudis.2006.11.004. PMID 17198969.
- ↑ Kucherenko, A. S.; Siyutkin, D. E.; Maltsev, O. V.; Kochetkov, S. V.; Zlotin, S. G. (2013). "Asymmetric organocatalysis: From proline to highly efficient immobilized organocatalysts". Russian Chemical Bulletin. 61 (7): 1313. doi:10.1007/s11172-012-0177-4.
- ↑ Ahrendt, K. A.; Borths, C. J.; MacMillan, D. W. C. (2000). "New Strategies for Organic Catalysis: The First Highly Enantioselective Organocatalytic Diels-Alder Reaction". J. Am. Chem. Soc. 122 (17): 4243–4244. doi:10.1021/ja000092s.
- ↑ Gérald Lelais; David W. C. MacMillan (2006). "Modern Strategies in Organic Catalysis: The Advent and Development of Iminium Activation" (PDF). Aldrichimica Acta. 39 (3): 79.
- ↑ Nis Halland; Tore Hansen; Karl Anker Jørgensen (2003). "Organocatalytic Asymmetric Michael Reaction of Cyclic 1,3-Dicarbonyl Compounds and α,β-Unsaturated Ketones - A Highly Atom-Economic Catalytic One-Step Formation of Optically Active Warfarin Anticoagulant". Angew. Chem. Int. Ed. 42 (40): 4955–4957. doi:10.1002/anie.200352136. PMID 14579449.
- ↑ Sandra Lee; David W. C. MacMillan (2007). "Organocatalytic Vinyl and Friedel-Crafts Alkylations with Trifluoroborate Salts". J. Am. Chem. Soc. 129 (50): 15438–15439. doi:10.1021/ja0767480. PMID 18031044.