Neurodegeneration
| Neurodegeneration | |
|---|---|
.gif) | |
| Para-sagittal MRI of the head in a patient with benign familial macrocephaly. | |
| Classification and external resources | |
| Specialty | neurology |
| ICD-10 | G30-G32 |
| MeSH | D019636 |
Neurodegeneration is the umbrella term for the progressive loss of structure or function of neurons, including death of neurons. Many neurodegenerative diseases including amyotrophic lateral sclerosis, Parkinson's, Alzheimer's, and Huntington's occur as a result of neurodegenerative processes. Such diseases are incurable, resulting in progressive degeneration and/or death of neuron cells.[1] As research progresses, many similarities appear that relate these diseases to one another on a sub-cellular level. Discovering these similarities offers hope for therapeutic advances that could ameliorate many diseases simultaneously. There are many parallels between different neurodegenerative disorders including atypical protein assemblies as well as induced cell death.[2][3] Neurodegeneration can be found in many different levels of neuronal circuitry ranging from molecular to systemic.
Links between neurodegenerative disorders
Genetics
Many neurodegenerative diseases are caused by genetic mutations, most of which are located in completely unrelated genes. In many of the different diseases, the mutated gene has a common feature: a repeat of the CAG nucleotide triplet. CAG encodes for the amino acid glutamine. A repeat of CAG results in a polyglutamine (polyQ) tract. Diseases showing this are known as polyglutamine diseases.[4][5]
- Polyglutamine: A repeat in this causes dominant pathogenesis. Extra glutamine residues can acquire toxic properties through a variety of ways, including irregular protein folding and degradation pathways, altered subcellular localization, and abnormal interactions with other cellular proteins.[4] PolyQ studies often use a variety of animal models because there is such a clearly defined trigger – repeat expansion. Extensive research has been done using the models of nematode (C. elegans), and fruit fly (Drosophila), mice, and non-human primates. Mammalian data is often needed for FDA approval of drugs, which means that the bulk of the research is done using mice. Using data from the other animals (C. elegans and Drosophila primarily) is often a precursor to finding the equivalent mammalian gene.[5][6]
- Nine inherited neurodegenerative diseases are caused by the expansion of the CAG trinucleotide and polyQ tract.[7] Two examples are Huntington's disease and spinocerebellar ataxias. For a complete list see the table under Polyglutamine (PolyQ) Diseases in the article Trinucleotide repeat disorder. While polyglutamine-repeat diseases encompass many different neurodegenerative disorders, there are many more it does not apply to. The genetics behind each disease are different and often unknown.
Protein misfolding
Several neurodegenerative diseases are classified as proteopathies as they are associated with the aggregation of misfolded proteins.
- alpha-synuclein: can aggregate to form insoluble fibrils in pathological conditions characterized by Lewy bodies, such as Parkinson's disease, dementia with Lewy bodies, and multiple system atrophy. Alpha-synuclein is the primary structural component of Lewy body fibrils. In addition, an alpha-synuclein fragment, known as the non-Abeta component (NAC), is found in amyloid plaques in Alzheimer's disease.
- tau: hyperphosphorylated tau protein is the main component of neurofibrillary tangles in Alzheimer's disease.
- beta amyloid: the major component of senile plaques in Alzheimer's disease.
- prion: main component of prion diseases and transmissible spongiform encephalopathies.
Intracellular mechanisms
Protein degradation pathways
Parkinson's disease and Huntington's disease are both late-onset and associated with the accumulation of intracellular toxic proteins. Diseases caused by the aggregation of proteins are known as proteinopathies, and they are primarily caused by aggregates in the following structures:[2]
- cytosol, e.g. Parkinson's & Huntington's
- nucleus, e.g. Spinocerebellar ataxia type 1 (SCA1)
- endoplasmic reticulum (ER), (as seen with neuroserpin mutations that cause familial encephalopathy with neuroserpin inclusion bodies)
- extracellularly excreted proteins, amyloid-β in Alzheimer's disease
There are two main avenues eukaryotic cells use to remove troublesome proteins or organelles:
- ubiquitin–proteasome: protein ubiquitin along with enzymes is key for the degradation of many proteins that cause proteinopathies including polyQ expansions and alpha-synucleins. Research indicates proteasome enzymes may not be able to correctly cleave these irregular proteins, which could possibly result in a more toxic species. This is the primary route cells use to degrade proteins.[2]
- Decreased proteasome activity is consistent with models in which intracellular protein aggregates form. It is still unknown whether or not these aggregates are a cause or a result of neurodegeneration.[2]
- autophagy–lysosome pathways: a form of programmed cell death (PCD), this becomes the favorable route when a protein is aggregate-prone meaning it is a poor proteasome substrate. This can be split into two forms of autophagy: macroautophagy and chaperone-mediated autophagy (CMA).[2]
- macroautophagy is involved with nutrient recycling of macromolecules under conditions of starvation, certain apoptotic pathways, and if absent, leads to the formation of ubiquinated inclusions. Experiments in mice with neuronally confined macroautophagy-gene knockouts develop intraneuronal aggregates leading to neurodegeneration.[2]
- chaperone-mediated autophagy defects may also lead to neurodegeneration. Research has shown that mutant proteins bind to the CMA-pathway receptors on lysosomal membrane and in doing so block their own degradation as well as the degradation of other substrates.[2]
Membrane damage
Damage to the membranes of organelles by monomeric or oligomeric proteins could also contribute to these diseases. Alpha-synuclein can damage membranes by inducing membrane curvature,[8] and cause extensive tubulation and vesiculation when incubated with artificial phospholipid vesicles.[8] The tubes formed from these lipid vesicles consist of both micellar as well as bilayer tubes. Extensive induction of membrane curvature is deleterious to the cell and would eventually lead to cell death.Apart from tubular structures, alpha-synuclein can also form lipoprotein nanoparticles similar to apolipoproteins.
Mitochondrial dysfunction
The most common form of cell death in neurodegeneration is through the intrinsic mitochondrial apoptotic pathway. This pathway controls the activation of caspase-9 by regulating the release of cytochrome c from the mitochondrial intermembrane space (IMS). Reactive oxygen species (ROS) are normal byproducts of mitochondrial respiratory chain activity. ROS concentration is mediated by mitochondrial antioxidants such as manganese superoxide dismutase (SOD2) and glutathione peroxidase. Over production of ROS (oxidative stress) is a central feature of all neurodegenerative disorders. In addition to the generation of ROS, mitochondria are also involved with life-sustaining functions including calcium homeostasis, PCD, mitochondrial fission and fusion, lipid concentration of the mitochondrial membranes, and the mitochondrial permeability transition. Mitochondrial disease leading to neurodegeneration is likely, at least on some level, to involve all of these functions.[9]
There is strong evidence that mitochondrial dysfunction and oxidative stress play a causal role in neurodegenerative disease pathogenesis, including in four of the more well known diseases Alzheimer's, Parkinson's, Huntington's, and Amyotrophic lateral sclerosis.[10]
Axonal transport
Axonal swelling and spheroids have been observed in many different neurodegenerative diseases. This suggests that defective axons are not only present in diseased neurons, but also that they may cause certain pathological insult due to accumulation of organelles. Axonal transport can be disrupted by a variety of mechanisms including damage to: kinesin and cytoplasmic dynein, microtubules, cargoes, and mitochondria.[11] When axonal transport is severely disrupted a degenerative pathway known as Wallerian-like degeneration is often triggered.[12]
Programmed cell death
Programmed cell death (PCD) is death of a cell in any form, mediated by an intracellular program.[13] This process can be activated in neurodegenerative diseases including Parkinson's disease, amytrophic lateral sclerosis, Alzheimer's disease and Huntington's disease.[14] There are, however, situations in which these mediated pathways are artificially stimulated due to injury or disease.[3]
Apoptosis (type I)
Apoptosis is a form of programmed cell death in multicellular organisms. It is one of the main types of programmed cell death (PCD) and involves a series of biochemical events leading to a characteristic cell morphology and death.
- Extrinsic apoptotic pathways: Occur when factors outside the cell activate cell surface death receptors (e.g., Fas) that result in the activation of caspases-8 or -10.[3]
- Intrinsic apoptotic pathways: Result from mitochondrial release of cytochrome c or endoplasmic reticulum malfunctions, each leading to the activation of caspase-9. The nucleus and Golgi apparatus are other organelles that have damage sensors, which can lead the cells down apoptotic pathways.[3][15]
Caspases (cysteine-aspartic acid proteases) cleave at very specific amino acid residues. There are two types of caspases: initiators and effectors. Initiator caspases cleave inactive forms of effector caspases. This activates the effectors that in turn cleave other proteins resulting in apoptotic initiation.[3]
Autophagic (type II)
Autophagy is essentially a form of intracellular phagocytosis in which a cell actively consumes damaged organelles or misfolded proteins by encapsulating them into an autophagosome, which fuses with a lysosome to destroy the contents of the autophagosome. Many neurodegenerative diseases show unusual protein aggregates. This could potentially be a result of underlying autophagic defect common to multiple neurodegenerative diseases. It is important to note that this is a hypothesis, and more research must be done.[3]
Cytoplasmic (type III)
The final and least understood PCD mechanism is through non-apoptotic processes. These fall under Type III, or cytoplasmic cell death. Many other forms of PCD are observed but not fully understood or accepted by the scientific community. For example, PCD might be caused by trophotoxicity, or hyperactivation of trophic factor receptors. In addition to this, other cytotoxins that induce PCD at low concentrations act to cause necrosis, or aponecrosis – the combination of apoptosis and necrosis, when in higher concentrations. It is still unclear exactly what combination of apoptosis, non-apoptosis, and necrosis causes different kinds of aponecrosis.[3]
PCD and neurodegeneration
In the above-mentioned neurodegenerative diseases, PCD may be pathogenic. In order to identify the potential of neuroprotective targets in PCD machinery, there were experimental models conducted on these neurodegenerative diseases. These studies showed that the expression of certain components have been altered by genetic and pharmacological means. Expression of PCD molecular components are said to be controlled by gene and antisense therapy, but this needs further research. Pharmacological approaches involve inhibitors of caspase activity, and caspase inhibition might delay cell death in the different experimental models.[14]
Specific disorders
Alzheimer's disease
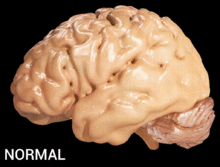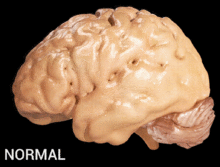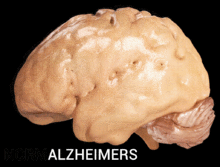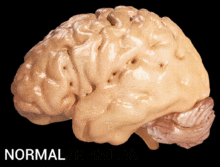
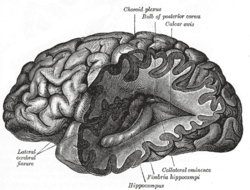
Alzheimer's disease is characterised by loss of neurons and synapses in the cerebral cortex and certain subcortical regions. This loss results in gross atrophy of the affected regions, including degeneration in the temporal lobe and parietal lobe, and parts of the frontal cortex and cingulate gyrus.[16]
Alzheimer's disease has been hypothesized to be a protein misfolding disease (proteopathy), caused by accumulation of abnormally folded A-beta and tau proteins in the brain.[17] Plaques are made up of small peptides, 39–43 amino acids in length, called beta-amyloid (also written as A-beta or Aβ). Beta-amyloid is a fragment from a larger protein called amyloid precursor protein (APP), a transmembrane protein that penetrates through the neuron's membrane. APP is critical to neuron growth, survival and post-injury repair.[18][19] In Alzheimer's disease, an unknown process causes APP to be divided into smaller fragments by enzymes through proteolysis.[20] One of these fragments gives rise to fibrils of beta-amyloid, which form clumps that deposit outside neurons in dense formations known as senile plaques.[21][22]
Parkinson's disease
Parkinson's disease is the second most common neurodegenerative disorder[23] and manifests as bradykinesia, rigidity, resting tremor and posture instability. The crude prevalence rate of PD has been reported to range from 15 per 100,000 to 12,500 per 100,000, and the incidence of PD from 15 per 100,000 to 328 per 100,000, with the disease being less common in Asian countries. Parkinson's disease is a degenerative disorder of the central nervous system. It results from the death of dopamine-generating cells in the substantia nigra, a region of the midbrain; the cause of cell-death is unknown. The following paragraph is an excerpt from the Pathophysiology section of the article Parkinson's disease.
The mechanism by which the brain cells in Parkinson's are lost may consist of an abnormal accumulation of the protein alpha-synuclein bound to ubiquitin in the damaged cells. The alpha-synuclein-ubiquitin complex cannot be directed to the proteosome. This protein accumulation forms proteinaceous cytoplasmic inclusions called Lewy bodies. The latest research on pathogenesis of disease has shown that the death of dopaminergic neurons by alpha-synuclein is due to a defect in the machinery that transports proteins between two major cellular organelles — the endoplasmic reticulum (ER) and the Golgi apparatus. Certain proteins like Rab1 may reverse this defect caused by alpha-synuclein in animal models.[24]
Recent research suggests that impaired axonal transport of alpha-synuclein leads to its accumulation in the Lewy bodies. Experiments have revealed reduced transport rates of both wild-type and two familial Parkinson's disease-associated mutant alpha-synucleins through axons of cultured neurons.[11] Membrane damage by alpha-synuclein could be another Parkinson's disease mechanism.[8]
The main known risk factor is age. Susceptibility genes including α-synuclein, leucine-rich repeat kinase 2 (LRRK-2), and glucocerebrosidase (GBA) have shown that genetic predisposition is another important causal factor.
Huntington's disease
The following paragraph is an excerpt from the Mechanism section of the article Huntington's disease.
HD causes astrogliosis[25] and loss of medium spiny neurons.[26][27] Areas of the brain are affected according to their structure and the types of neurons they contain, reducing in size as they cumulatively lose cells. The areas affected are mainly in the striatum, but also the frontal and temporal cortices.[28] The striatum's subthalamic nuclei send control signals to the globus pallidus, which initiates and modulates motion. The weaker signals from subthalamic nuclei thus cause reduced initiation and modulation of movement, resulting in the characteristic movements of the disorder, notably chorea.[29]
Mutant Huntingtin is an aggregate-prone protein. During the cells' natural clearance process, these proteins are retrogradely transported to the cell body for destruction by lysosomes. It is a possibility that these mutant protein aggregates damage the retrograde transport of important cargoes such as BDNF by damaging molecular motors as well as microtubules.[11]
Amyotrophic lateral sclerosis (ALS)
Amyotrophic lateral sclerosis (ALS or Lou Gehrig’s Disease) is a disease in which motor neurons are selectively targeted for degeneration. In 1993, missense mutations in the gene encoding the antioxidant enzyme Cu/Zn superoxide dismutase 1 (SOD1) were discovered in subsets of patients with familial ALS. This discovery led researchers to focus on unlocking the mechanisms for SOD1-mediated diseases. However, the pathogenic mechanism underlying SOD1 mutant toxicity has yet to be resolved. More recently, TDP-43 and FUS protein aggregates have been implicated in some cases of the disease, and a mutation in chromosome 9 (C9orf72) is thought to be the most common known cause of sporadic ALS.
Recent independent research by Nagai et al.[30] and Di Giorgio et al.[31] provide in vitro evidence that the primary cellular sites where SOD1 mutations act are located on astrocytes. Astrocytes then cause the toxic effects on the motor neurons. The specific mechanism of toxicity still needs to be investigated, but the findings are significant because they implicate cells other than neuron cells in neurodegeneration.[32]
Batten disease
Batten disease is a rare and fatal recessive neurodegenerative disorder that begins at birth.
Aging and neurodegeneration
The greatest risk factor for neurodegenerative diseases is aging. Mitochondrial DNA mutations as well as oxidative stress both contribute to aging.[10] Many of these diseases are late-onset, meaning there is some factor that changes as a person ages for each disease.[2] One constant factor is that in each disease, neurons gradually lose function as the disease progresses with age.
Therapeutics
The process of neurodegeneration is not well-understood so the diseases that stem from it have, as yet, no cures. In the search for effective treatments (as opposed to palliative care), investigators employ animal models of disease to test potential therapeutic agents. Model organisms provide an inexpensive and relatively quick means to perform two main functions: target identification and target validation. Together, these help show the value of any specific therapeutic strategies and drugs when attempting to ameliorate disease severity. An example is the drug Dimebon (Medivation). This drug is in phase III clinical trials for use in Alzheimer's disease, and also recently finished phase II clinical trials for use in Huntington's disease.[5] In March 2010, the results of a clinical trial phase III were released; the investigational Alzheimer's disease drug Dimebon failed in the pivotal CONNECTION trial of patients with mild-to-moderate disease.[33] With CONCERT, the remaining Pfizer and Medivation Phase III trial for Dimebon (latrepirdine) in Alzheimer's disease failed in 2012, effectively ending the development in this indication.[34]
In another experiment using a rat model of Alzheimer's disease, it was demonstrated that systemic administration of hypothalamic proline-rich peptide (PRP)-1 offers neuroprotective effects and can prevent neurodegeneration in hippocampus amyloid-beta 25–35. This suggests that there could be therapeutic value to PRP-1.[35]
Protein degradation offers therapeutic options both in preventing the synthesis and degradation of irregular proteins. There is also interest in upregulating autophagy to help clear protein aggregates implicated in neurodegeneration. Both of these options involve very complex pathways that we are only beginning to understand.[2]
The goal of immunotherapy is to enhance aspects of the immune system. Both active and passive vaccinations have been proposed for Alzheimer's disease and other conditions, however more research must be done to prove safety and efficacy in humans.[36]
See also
- Trinucleotide repeat disorders
- Dementia
- Prevention of dementia
- Proteopathy
- Nervous system
- Amyloids
- JUNQ and IPOD
References
- ↑ "What is Neurodegenerative Disease?". JPND Research. JPND Research. Retrieved February 7, 2015.
- 1 2 3 4 5 6 7 8 9 Rubinsztein DC (October 2006). "The roles of intracellular protein-degradation pathways in neurodegeneration". Nature. 443 (7113): 780–6. Bibcode:2006Natur.443..780R. doi:10.1038/nature05291. PMID 17051204.
- 1 2 3 4 5 6 7 Bredesen DE, Rao RV, Mehlen P (October 2006). "Cell death in the nervous system". Nature. 443 (7113): 796–802. Bibcode:2006Natur.443..796B. doi:10.1038/nature05293. PMID 17051206.
- 1 2 Thompson LM (April 2008). "Neurodegeneration: a question of balance". Nature. 452 (7188): 707–8. Bibcode:2008Natur.452..707T. doi:10.1038/452707a. PMID 18401401.
- 1 2 3 Marsh JL, Lukacsovich T, Thompson LM (March 2009). "Animal Models of Polyglutamine Diseases and Therapeutic Approaches". The Journal of Biological Chemistry. 284 (12): 7431–5. doi:10.1074/jbc.R800065200. PMC 2658038
 . PMID 18957429.
. PMID 18957429. - ↑ Orr HT (March 2009). "Unstable Nucleotide Repeat Minireview Series: A Molecular Biography of Unstable Repeat Disorders". The Journal of Biological Chemistry. 284 (12): 7405. doi:10.1074/jbc.R800067200. PMC 2658033. PMID 18957428.
- ↑ Zoghbi HY, Orr HT (March 2009). "Pathogenic Mechanisms of a Polyglutamine-mediated Neurodegenerative Disease, Spinocerebellar Ataxia Type 1". The Journal of Biological Chemistry. 284 (12): 7425–9. doi:10.1074/jbc.R800041200. PMC 2658037. PMID 18957430.
- 1 2 3 Varkey J, Isas JM, Mizuno N, et al. (October 2010). "Membrane curvature induction and tubulation are common features of synucleins and apolipoproteins". The Journal of Biological Chemistry. 285 (42): 32486–93. doi:10.1074/jbc.M110.139576. PMC 2952250. PMID 20693280.
- ↑ DiMauro S, Schon EA (2008). "Mitochondrial disorders in the nervous system". Annual Review of Neuroscience. 31: 91–123. doi:10.1146/annurev.neuro.30.051606.094302. PMID 18333761.
- 1 2 Lin MT, Beal MF (October 2006). "Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases". Nature. 443 (7113): 787–95. Bibcode:2006Natur.443..787L. doi:10.1038/nature05292. PMID 17051205.
- 1 2 3 De Vos KJ, Grierson AJ, Ackerley S, Miller CC (2008). "Role of axonal transport in neurodegenerative diseases". Annual Review of Neuroscience. 31: 151–73. doi:10.1146/annurev.neuro.31.061307.090711. PMID 18558852.
- ↑ Coleman MP & Freeman MF 'Wallerian degeneration, WldS and Nmnat' Annual Review of Neuroscience 2010, 33: 245-67
- ↑ Engelberg-Kulka H, Amitai S, Kolodkin-Gal I, Hazan R (October 2006). "Bacterial Programmed Cell Death and Multicellular Behavior in Bacteria". PLoS Genetics. 2 (10): e135. doi:10.1371/journal.pgen.0020135. PMC 1626106. PMID 17069462.
- 1 2 Vila, Miquel; Przedbroski, Serge (May 2003). "Targeting Programmed Cell Death in Neurodegenrative Diseases". Nature Reviews. 4: 1–11.
- ↑ Green DR, Kroemer G (October 2005). "Pharmacological manipulation of cell death: clinical applications in sight?". The Journal of Clinical Investigation. 115 (10): 2610–7. doi:10.1172/JCI26321. PMC 1236695. PMID 16200193.
- ↑ Wenk GL (2003). "Neuropathologic changes in Alzheimer's disease". J Clin Psychiatry. 64 Suppl 9: 7–10. PMID 12934968.
- ↑ Hashimoto M, Rockenstein E, Crews L, Masliah E (2003). "Role of protein aggregation in mitochondrial dysfunction and neurodegeneration in Alzheimer's and Parkinson's diseases". Neuromolecular Med. 4 (1–2): 21–36. doi:10.1385/NMM:4:1-2:21. PMID 14528050.
- ↑ Priller C, Bauer T, Mitteregger G, Krebs B, Kretzschmar HA, Herms J (July 2006). "Synapse formation and function is modulated by the amyloid precursor protein". J. Neurosci. 26 (27): 7212–21. doi:10.1523/JNEUROSCI.1450-06.2006. PMID 16822978.
- ↑ Turner PR, O'Connor K, Tate WP, Abraham WC (May 2003). "Roles of amyloid precursor protein and its fragments in regulating neural activity, plasticity and memory". Prog. Neurobiol. 70 (1): 1–32. doi:10.1016/S0301-0082(03)00089-3. PMID 12927332.
- ↑ Hooper NM (April 2005). "Roles of proteolysis and lipid rafts in the processing of the amyloid precursor protein and prion protein". Biochem. Soc. Trans. 33 (Pt 2): 335–8. doi:10.1042/BST0330335. PMID 15787600.
- ↑ Tiraboschi P, Hansen LA, Thal LJ, Corey-Bloom J (June 2004). "The importance of neuritic plaques and tangles to the development and evolution of AD". Neurology. 62 (11): 1984–9. doi:10.1212/01.WNL.0000129697.01779.0A. PMID 15184601.
- ↑ Ohnishi S, Takano K (March 2004). "Amyloid fibrils from the viewpoint of protein folding". Cell. Mol. Life Sci. 61 (5): 511–24. doi:10.1007/s00018-003-3264-8. PMID 15004691.
- ↑ Epidemiology of Parkinson's disease.
- ↑ "Parkinson's Disease Mechanism Discovered," HHMI Research News June 22, 2006.
- ↑ Lobsiger CS, Cleveland DW (November 2007). "Glial cells as intrinsic components of non-cell autonomous neurodegenerative disease". Nat. Neurosci. 10 (11): 1355–60. doi:10.1038/nn1988. PMC 3110080. PMID 17965655.
- ↑ Purves D, Augustine GA, Fitzpatrick D, Hall W, LaMantia AS, McNamara JO, Williams SM (2001). "Modulation of Movement by the Basal Ganglia - Circuits within the Basal Ganglia System". In Dale Purves. Neuroscience (2nd ed.). Sunderland, MA: Sinauer Associates. ISBN 0-87893-742-0.
- ↑ Estrada Sánchez AM, Mejía-Toiber J, Massieu L (April 2008). "Excitotoxic neuronal death and the pathogenesis of Huntington's disease". Arch. Med. Res. 39 (3): 265–76. doi:10.1016/j.arcmed.2007.11.011. PMID 18279698.
- ↑ Purves D, Augustine GA, Fitzpatrick D, Hall W, LaMantia AS, McNamara JO, Williams SM (2001). "Modulation of Movement by the Basal Ganglia - Box A. Huntington's Disease". In Dale Purves. Neuroscience (2nd ed.). Sunderland, MA: Sinauer Associates. ISBN 0-87893-742-0.
- ↑ Crossman AR (May 2000). "Functional anatomy of movement disorders" (PDF). J. Anat. 196 (4): 519–25. doi:10.1046/j.1469-7580.2000.19640519.x. PMC 1468094. PMID 10923984.
- ↑ Nagai M, Re DB, Nagata T, et al. (May 2007). "Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons". Nature Neuroscience. 10 (5): 615–22. doi:10.1038/nn1876. PMID 17435755.
- ↑ Di Giorgio FP, Carrasco MA, Siao MC, Maniatis T, Eggan K (May 2007). "Non–cell autonomous effect of glia on motor neurons in an embryonic stem cell–based ALS model". Nature Neuroscience. 10 (5): 608–14. doi:10.1038/nn1885. PMC 3139463. PMID 17435754.
- ↑ Julien JP (May 2007). "ALS: astrocytes move in as deadly neighbors". Nature Neuroscience. 10 (5): 535–7. doi:10.1038/nn0507-535. PMID 17453052.
- ↑ Dimebon Disappoints in Phase 3 Trial
- ↑ Sweetlove M: Phase III CONCERT Trial of Latrepirdine. Negative results. Pharm Med 2012;26(2):113-115
- ↑ Galoyan AA, Sarkissian JS, Chavushyan VA, et al. (September 2008). "Neuroprotection by hypothalamic peptide proline-rich peptide-1 in Aβ25–35 model of Alzheimer's disease". Alzheimer's & Dementia. 4 (5): 332–44. doi:10.1016/j.jalz.2007.10.019. PMID 18790460.
- ↑ Brody DL, Holtzman DM (2008). "Active and Passive Immunotherapy for Neurodegenerative Disorders". Annual Review of Neuroscience. 31: 175–93. doi:10.1146/annurev.neuro.31.060407.125529. PMC 2561172. PMID 18352830.