Mycoplasma pneumoniae
| Mycoplasma pneumoniae | |
|---|---|
| Scientific classification | |
| Kingdom: | Bacteria |
| Division: | Tenericutes |
| Class: | Mollicutes |
| Order: | Mycoplasmatales |
| Family: | Mycoplasmataceae |
| Genus: | Mycoplasma |
| Binomial name | |
| Mycoplasma pneumoniae Somerson et al., 1963 | |
Mycoplasma pneumoniae is a very small bacterium in the class Mollicutes. It is a human pathogen that causes the disease mycoplasma pneumonia, a form of atypical bacterial pneumonia related to cold agglutinin disease. M. pneumoniae is characterized by the absence of a peptidoglycan cell wall and resulting resistance to many antibacterial agents. The persistence of M. pneumoniae infections even after treatment is associated with its ability to mimic host cell surface composition.
Discovery and history
In 1898, Nocard and Roux were the first to isolate a mycoplasma species in culture from bovine, however it wasn't until 1944 when Mycoplasma pneumoniae, known then as Eaton agent or Eaton's agent,[1] was isolated and described from a patient with primary atypical pneumonia.[2]
Initially M. pneumoniae was considered as a virus rather than a bacterium, when Eaton and colleagues cultured the causative agent of human primary atypical pneumonia (PAP) or "walking pneumonia".[3] The terms 'walking pneumonia' and 'atypical pneumonia' were coined to describe the unresponsiveness of pneumonia inducing M. pneumoniae infections to antibiotics like penicillin.[2] Eaton's agent could be grown in chicken embryos and passed through a filter that excluded normal bacteria. Eaton suggested the possibility that the disease was caused by a mycoplasma, but the agent did not grow on the standard pleuropneumonia-like organism (PPLO) media of the time. These observations led to the conclusion that the causative agent of PAP was a virus. Researchers at that time showed that the cultured agent could induce disease in experimentally infected cotton rats and hamsters. In spite of controversy whether the researchers had truly isolated the causative agent of PAP (based largely on the unusual immunological response of patients with PAP), in retrospect their evidence along with that of colleagues and competitors appears to have been quite conclusive.[4] There were reports linking Eaton agent to the PPLOs or mycoplasmas, well known then as parasites of cattle and rodents, due to sensitivity to antimicrobials. Studies that followed until 1963 determined that Eaton’s agent was a bacterium that caused human lower respiratory tract infections.[2]
Taxonomy and classification
The term mycoplasma (“mykes”, meaning fungus and “plasma”, meaning formed) is derived from the fungal-like growth of some mycoplasma species.[2] The mycoplasmas were classified as Mollicutes (“mollis”, meaning soft and “cutis”, meaning skin) in 1960 due to their small size and genome, lack of cell wall, low G+C content and unusual nutritional needs.[2][5] M. pneumoniae has also been designated as an arginine nonfermenting species.[6] Mycoplasmas are further classified by the sequence composition of 16s rRNA. All mycoplasmas of the pneumoniae group possess similar 16s rRNA variations unique to the group, of which M. pneumoniae has a 6.3% variation in the conserved regions, that suggest mycoplasmas formed by degenerative evolution from the gram-positive eubacterial group that includes bacilli, streptococci, and lactobacilli.[2][5][6] M. pneumoniae is a member of the Mycoplasmataceae family and Mycoplasmatales order.[2]
Cell biology
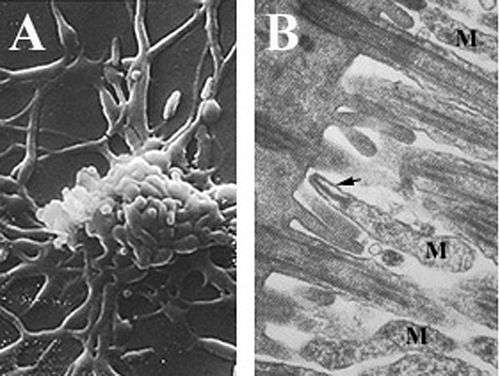
Mycoplasmas, are among the smallest self-replicating organisms, are parasitic species that lack a cell wall and periplasmic space, have reduced genomes, and limited metabolic activity.[2][6][7] Mycoplasma pneumoniae cells have an elongated shape that is approximately 0.1–0.2 µm (100-200 nm) in width and 1-2 µm (1000-2000 nm) in length. The extremely small cell size means they are incapable of being examined by light microscopy; a stereomicroscope is required for viewing the morphology of M. pneumoniae colonies, which are usually less than 100 µm in length.[2] The inability to synthesize a peptidoglycan cell wall is due to the absence of genes encoding its formation and results in an increased importance in maintenance of osmotic stability to avoid desiccation.[2] The lack of a cell wall also calls for increased support of the cell membrane(reinforced with sterols), which includes a rigid cytoskeleton composed of an intricate protein network and, potentially, an extracellular capsule to facilitate adherence to the host cell.[2] M. pneumoniae are the only bacterial cells that possess cholesterol in their cell membrane (obtained from the host) and possess more genes that encode for membrane lipoprotein variations than other mycoplasmas,[6] which are thought to be associated with its parasitic lifestyle. M. pneumoniae cells also possess an attachment organelle, which is used in the gliding motility of the organism by an unknown mechanism.[2]
Genomics and metabolic reconstruction
Sequencing of the M. pneumoniae genome in 1996 revealed it is 816,394 bp in size.[5] The genome contains 687 genes that encode for proteins, of which about 56.6% code for essential metabolic enzymes; notably those involved in glycolysis and organic acid fermentation.[2][5][6][8] M. pneumoniae is consequently very susceptible to loss of enzymatic function by gene mutations, as the only buffering systems against functional loss by point mutations are for maintenance of the pentose phosphate pathway and nucleotide metabolism.[8] Loss of function in other pathways is suggested to be compensated by host cell metabolism.[8] In addition to the potential for loss of pathway function, the reduced genome of M. pneumoniae outright lacks a number of pathways, including the TCA cycle, respiratory electron transport chain, and biosynthesis pathways for amino acids, fatty acids, cholesterol and purines and pyrimidines.[2][6][8] These limitations make M. pneumoniae dependent upon import systems to acquire essential building blocks from their host or the environment that cannot be obtained through glycolytic pathways.[6][8] Along with energy costly protein and RNA production, a large portion of energy metabolism is exerted to maintain proton gradients (up to 80%) due to the high surface area to volume ratio of M. pneumoniae cells. Only 12 – 29% of energy metabolism is directed at cell growth, which is unusually low for bacterial cells, and is thought to be an adaptation of its parasitic lifestyle.[8] Unlike other bacteria, M. pneumoniae uses the codon UGA to code for tryptophan rather than using it as a stop codon.[2][5]
Host and reproduction
Mycoplasma pneumoniae exclusively parasitizes humans, and has never been isolated as a free-living organism due to its dependence upon the host for survival. Reproduction, therefore, is dependent upon attachment to a host cell. According to Waite and Talkington, specialized reproduction occurs by “binary fission, temporally linked with duplication of its attachment organelle, which migrates to the opposite pole of the cell during replication and before nucleoid separation”.[2] Mutations that affect the formation of the attachment organelle not only hinder motility and cell division, but also reduce the ability of M. pneumoniae cells to adhere to the host cell.[6]
Pathogenicity
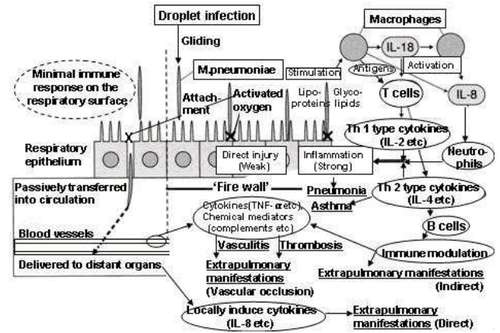
Mycoplasma pneumoniae parasitizes the respiratory tract epithelium of humans.[2] Adherence to the respiratory epithelial cells is thought to occur via the attachment organelle, followed by evasion of host immune system by intracellular localization and adjustment of the cell membrane composition to mimic the host cell membrane.
Cytadherence
Adherence of M. pneumoniae to a host cell (usually a respiratory tract cell, but occasionally an erythrocyte or urogenital lining cell) is the initiating event for pneumonic disease and related symptoms.[2] The specialized attachment organelle is a polar, electron dense and elongated cell extension that facilitates motility and cytadherence to host cells.[2][6] It is composed of a central filament surrounded by an intracytoplasmic space, along with a number of adhesins and structural and accessory proteins localized at the tip of the organelle.[2][6] A variety of proteins are known to contribute to the formation and functionality of the attachment organelle, including the accessory proteins HMW1–HMW5, P30, P56, and P90 that confer structure and adhesin support, and P1, P30 and P116 which are involved directly in attachment.[2][9][10] This network of proteins participates not only in the initiation of attachment organelle formation and adhesion but also in motility.[10] The P1 adhesin (trypsin-sensitive protein) is a 120 kDa protein highly clustered on the surface of the attachment organelle tip in virulent mycoplasmas.[2][10][11] Both the presence of P1 and its concentration on the cell surface are required for the attachment of M. pneumoniae to the host cell. M. pneumoniae cells treated with monoclonal antibodies specific to the immunogenic C-terminus of the P1 adhesin have been shown to be inhibited in their ability to attach to the host cell surface by approximately 75%, suggesting P1 is a major component in cytadherence.[2][9][10] These antibodies also decreased the ability of the cell to glide quickly, which may contribute to decreased adherence to the host by hindering their capacity to locate a host cell.[9] Furthermore, mutations in P1 or degradation by trypsin treatment yield avirulent M. pneumoniae cells.[2] Loss of proteins in the cytoskeleton involved in the localization of P1 in the tip structure, such as HMW1–HMW3, also cause avirulence due to the lack of adhesin clustering.[10][11] Another protein considered to play an important role in cytadherence is P30, as M. pneumoniae cells with mutations in this protein or that have had antibodies raised against P30 are incapable of adhering to host cells.[2][6] P30 is not involved in the localization of P1 in the tip structure since P1 is trafficked to the attachment organelle in P30 mutants, but rather it may function as a receptor-binding accessory adhesin.[6][11] P30 mutants also display distinct morphological features such as multiple lobes and a rounded shape as opposed to elongated, which suggests P30 may interact with the cytoskeleton during formation of the attachment organelle.[6] A number of eukaryotic cell surface components have been implicated in the adherence of M. pneumoniae cells to the respiratory tract epithelium. Among them are sialoglycoconjugates, sulfated glycolipids, glycoproteins, fibronectin, and neuraminic acid receptors.[2][9][12] Lectins on the surface of the bacterial cells are capable of binding oligosaccharide chains on glycolipids and glycoproteins to facilitate attachment, in addition to the proteins TU and pyruvate dehydrogenase E1 β, which bind to fibronectin.[2][9]

Intracellular localization
Mycoplasma pneumoniae is known to evade host immune system detection, resist antibiotic treatment, and cross mucosal barriers, which may be due to its ability to fuse with host cells and survive intracellularly.[2][7] In addition to the close physical proximity of M. pneumoniae and host cells, the lack of cell wall and peculiar cell membrane components, like cholesterol, may facilitate fusion (1). Internal localization may produce chronic or latent infections as M. pneumoniae is capable of persisting, synthesizing DNA, and replicating within the host cell even after treatment with antibiotics.[7] The exact mechanism of intracellular localization is unknown, however the potential for cytoplasmic sequestration within the host explains the difficulty in completely eliminating M. pneumoniae infections in afflicted individuals.[2]
Immune response
In addition to evasion of host immune system by intracellular localization, M. pneumoniae can change the composition of its cell membrane to mimic the host cell membrane and avoid detection by immune system cells. M. pneumoniae cells possess a number of protein and glycolipid antigens that elicit immune responses, but variation of these surface antigens would allow the infection to persist long enough for M. pneumoniae cells to fuse with host cells and escape detection. The similarity between the compositions of M. pneumoniae and human cell membranes can also result in autoimmune responses in several organs and tissues.[2]
Cytotoxicity and organismal effects
The main cytotoxic effect of M. pneumoniae is local disruption of tissue and cell structure along the respiratory tract epithelium due to its close proximity to host cells. Attachment of the bacteria to host cells can result in loss of cilia, a reduction in metabolism, biosynthesis, and import of macromolecules, and, eventually, infected cells may be shed from the epithelial lining.[2] M. pneumoniae produces a unique virulence factor known as Community Acquired Respiratory Distress Syndrome (CARDS) toxin.[13] The CARDS toxin most likely aids in the colonization and pathogenic pathways of M. pneumoniae, leading to inflammation and airway dysfunction. In addition, the formation of hydrogen peroxide is a key virulence factor in M. pneumoniae infections.[2] Attachment of M. pneumoniae to erythrocytes permits diffusion of hydrogen peroxide from the bacteria to the host cell without detoxification by catalase or peroxidase, which can injure the host cell by reducing glutathione, damaging lipid membranes and causing protein denaturation.[2][12] Local damage may also be a result of lactoferrin acquisition and subsequent hydroxyl radical, superoxide anion and peroxide formation.[2] The cytotoxic effects of M. pneumoniae infections translate into common symptoms like coughing and lung irritation that may persist for months after infection has subsided. Local inflammation and hyperresponsiveness by infection induced cytokine production has been associated with chronic conditions such as bronchial asthma and has also been linked to progression of symptoms in individuals with cystic fibrosis and COPD.[2]
Epidemiology
The incidence of disease does not appear be related to season or geography, however infection tends to occur more frequently during the summer and fall months when other respiratory pathogens are less prevalent. Reinfection and epidemic cycling is thought to be a result of P1 adhesin subtype variation.[2] Approximately 40% of community-acquired pneumonia is due to M. pneumoniae infections, with children and elderly individuals being most susceptible, however no personal risk factors for acquiring M. pneumoniae induced pneumonia have been determined.[2][14] Transmission of M. pneumoniae can only occur through close contact and exchange of aerosols by coughing due to the increased susceptibility of the cell wall-lacking organism to desiccation. Outbreaks of M. pneumoniae infections tend to occur within groups of people in close and prolonged proximity, including schools, institutions, military bases, and households.[2]
Symptoms of infection
M. pneumoniae is known to cause a host of symptoms such as primary atypical pneumonia, tracheobronchitis, and upper respiratory tract disease. Primary atypical pneumonia is one of the most severe types of manifestation, with tracheobronchitis being the most common symptom and another 15% of cases, usually adults, remain asymptomatic.[2][14] Symptomatic infections tend to develop over a period of several days and manifestation of pneumonia can be confused with a number of other bacterial pathogens and conditions that cause pneumonia. Tracheobronchitis is most common in children due to a reduced immune system capacity, and up to 18% of infected children require hospitalization.[2] Common mild symptoms include sore throat, wheezing and coughing, fever, headache, coryza, myalgia and feelings of unease, in which symptom intensity and duration can be limited by early treatment with antibiotics. Rarely, M. pneumoniae pneumonia results in death due to lesions and ulceration of the epithelial lining, pulmonary edema, and bronchiolitis obliterans. Extrapulmonary symptoms such as autoimmune responses, central nervous system complications, and dermatological disorders have been associated with M. pneumoniae infections in up to 25% of cases.[2]
Diagnosis
Diagnosis of Mycoplasma pneumoniae infections is complicated by the delayed onset of symptoms and the similarity of symptoms to other pulmonary conditions. Often, M. pneumoniae infections are diagnosed as other conditions and, occasionally, non-pathogenic mycoplasmas present in the respiratory tract are mistaken for M. pneumoniae.[2] Historically, diagnosis of M. pneumoniae infections was made based on the presence of cold agglutinins and the ability of the infected material to reduce tetrazolium. Causative diagnosis is dependent upon laboratory testing, however these methods are more practical in epidemiological studies than in patient diagnosis.[2] Culture tests are rarely used as diagnosic tools; rather immunoblotting, immunofluorescent staining, hemadsorption tests, tetrazolium reduction, metabolic inhibition tests, serological assays, and polymerase chain reaction (PCR) are used for diagnosis and characterization of bacterial pneumonic infections.[2] PCR is the most rapid and effective way to determine the presence of M. pneumoniae, however the procedure does not indicate the activity or viability of the cells present.[14] EIA serological assays are the most common method of M. pneumoniae detection used in patient diagnosis due to the low cost and relatively short testing time. One drawback of serology is that viable organisms are required, which may overstate the severity of infection.[2] Neither of these methods, along with others, has been available to medical professionals in a rapid, efficient and inexpensive enough form to be used in routine diagnosis, leading to decreased ability of physicians to diagnose M. pneumoniae infections.
Treatment and prevention
The difficulty in eradicating Mycoplasma pneumoniae infections is due to the ability of the bacterium to persist within an individual, as well as the lack of cell wall in M. pneumoniae, which renders multiple antibiotics directed at the bacterial cell wall ineffective in treating infections.[2] M. pneumoniae therefore displays resistance to antimicrobials such as β-lactams, glycopeptides, sulfonamides, trimethoprim, polymixins, nalidixic acid, and rifampin.[2][14] The majority of antibiotics used to treat M. pneumoniae infections are targeted at bacterial rRNA in ribosomal complexes, including macrolides, tetracycline, ketolides, and fluoroquinolone, many of which can be administered orally.[2][15] Macrolides are capable of reducing hyperresponsiveness and protecting the epithelial lining from oxidative and structural damage, however they are capable only of inhibiting bacteria (bacteriostatic) and are not able to cause bacterial cell death.[2][7] The most common macrolides used in the treatment of infected children in Japan are erythromycin and clarithromycin, which inhibit bacterial protein synthesis by binding 23S rRNA.[15] Administration of antibiotics has been proven to reduce the longevity and intensity of M. pneumoniae infections in comparison to cases left untreated. Additionally, some high-dose steroid therapies have shown to reverse neurological effects in children with complicated infections.[2] Antimicrobial drug resistance rates for Mycoplasma pneumoniae were determined in clinical specimens and isolates obtained during 2011–2012 in Ontario, Canada. Of 91 M. pneumoniae drug-resistant specimens, 11 (12.1%) carried nucleotide mutations associated with macrolide resistance in the 23S rRNA gene. None of the M. pneumoniae specimens were resistant to fluoroquinolones or tetracyclines.[16]
Vaccine design for M. pneumoniae has been focused primarily on prevention of host cell attachment, which would prevent initiation of cytotoxicity and subsequent symptoms.[2] To date, vaccines targeted at the P1 adhesin have shown no reduction in the onset of infection, and some vaccine trials resulted in worsened symptoms due to immune system sensitization.[2] Introduction of peptides that block adhesion receptors on the surface of the host cell may also be able to prevent attachment of M. pneumoniae.[9]
Transmission of Mycoplasma pneumoniae infections is difficult to limit because of the several day period of infection before symptoms appear.[17] The lack of proper diagnostic tools and effective treatment for the bacterium also contribute to the outbreak of infection.[17] Using network theory, Meyers et al. analyzed the transmission of M. pneumoniae infections and developed control strategies based on the created model. They determined that cohorting is less effective due to the long incubation period, and so the best method of prevention is to limit caregiver-patient interactions and reduce the movement of caregivers to multiple wards.[17]
See also
References
- ↑ A.S. Dajani, W.A. Clyde Jr. and F.W. Denny (1965). "Experimental Infection with Mycoplasma Pneumoniae (Eaton's Agent)". The Journal of Experimental Medicine. 121 (6): 1071–1086. doi:10.1084/jem.121.6.1071. PMC 2138014
 . PMID 14319403.
. PMID 14319403. - 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 Ken; Waites, B; Deborah, F. Talkington (2004). "Mycoplasma pneumoniae and Its Role as a Human Pathogen". Clin. Microbiol. Rev. 17 (4): 697–728. doi:10.1128/CMR.17.4.697-728.2004.
- ↑ Eaton MD, Meiklejohn G, van Herrick W, Corey M (1945). "STUDIES ON THE ETIOLOGY OF PRIMARY ATYPICAL PNEUMONIA : III. SPECIFIC NEUTRALIZATION OF THE VIRUS BY HUMAN SERUM". J. Exp. Med. 82 (5): 329–42. doi:10.1084/jem.82.5.329. PMC 2135563. PMID 19871504.
- ↑ B.P. Marmion (1990). "Eaton agent—science and scientific acceptance: a historical commentary". Rev. Infect. Dis. 12 (2): 338–53. doi:10.1093/clinids/12.2.338. PMID 2109871.
- 1 2 3 4 5 Weisburg, W. G.; Tully, J. G.; Rose, D. L.; Pretzel, J.P.; Oyaizu, H.; Yang, D.; Mandelco, L.; Sechrest, J.; Lawrence, T. G.; Etten, J. Van (1989). "A phylogenetic analysis of the mycoplasmas: basis for their classification". J. Bacteriol. 171: 6455–6467.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 Romero-Arroyo, C. E.; Jordan, J.; Peacock, S. J.; Willby, M. J.; Farmer, M. A.; Krause, D. C. (1994). "Mycoplasma pneumoniae protein P30 is required for cytadherence and associated with proper cell development". J. Bacteriol. 181: 1079–1087. doi:10.1128/CMR.17.4.697-728.2004.
- 1 2 3 4 Dallo, S.; Baseman, J. (2000). "Intracellular DNA replication and long-term survival of pathogenic mycoplasmas". Microb. Pathog. 29: 301–309. doi:10.1006/mpat.2000.0395.
- 1 2 3 4 5 6 Wodke, J. A. H.; Puchałka, J.; Lluch-Senar, M.; Marcos, J.; Yus, E.; Godinho, M.; Gutiérrez-Gallego, R.; Serrano, L.; Klipp, E.; Maier, T. "Dissecting the energy metabolism in Mycoplasma pneumoniae through genome-scale metabolic modeling". Mol. Syst. Biol. 2010: 9. doi:10.1038/msb.2013.6.
- 1 2 3 4 5 6 Drasbek, M.; Christiansen, G.; Drasbek, K. R.; Holm, A.; Birkelund, S. (2007). "Interaction between the P1 protein of Mycoplasma pneumoniae and receptors on HEp-2 cells". Microbiology. 153: 3791–3799. doi:10.1099/mic.0.2007/010736-0.
- 1 2 3 4 5 Baseman, J. B.; Cole, R. M.; Krause, D. C.; Leith, D. K. (1982). "Molecular basis for cytadsorption of Mycoplasma pneumoniae". J. Bacteriol. 151: 1514–1522.
- 1 2 3 Hahn, T.-W; Willby, M. J.; Krause, D.C. (1998). "HMW1 Is Required for Cytadhesin P1 Trafficking to the Attachment Organelle in Mycoplasma pneumoniae". J. Bacteriol. 180: 1270–1276.
- 1 2 Sobeslavsky, B. Prescott; Chanock, R. M. "Adsorption of Mycoplasma pneumoniae to Neuraminic Acid Receptors of Various Cells and Possible Role in Virulence". J. Bacteriol. 96: 695–705.
- ↑ "CDC Mycoplasma Pneumoniae". CDC. CDC. Retrieved 23 September 2015.
- 1 2 3 4 Daxboeck, F.; Krause, R.; Wenisch, C. (2003). "Laboratory diagnosis of Mycoplasma pneumoniae infection". Clin. Microbiol. Infect. 9: 263–273. doi:10.1046/j.1469-0691.2003.00590.x.
- 1 2 Matsuoka, M.; Narita, M.; Okazaki, N.; Ohya, H.; Yamazaki, T.; Ouchi, K.; Suzuki, I.; Andoh, T.; Kenri, T.; Sasaki, Y.; Horino, A.; Shintani, M.; Arakawa, Y.; Sasaki, T. (2004). "Characterization and Molecular Analysis of Macrolide-Resistant Mycoplasma pneumoniae Clinical Isolates Obtained in Japan". Antimicrob. Agents Chemother. 48: 4624–4630. doi:10.1128/AAC.48.12.4624-4630.2004.
- ↑ Eshaghi, A; Memari, N; Tang, P; Olsha, R; Farrell, DJ; Low, DE; et al. "Macrolide-resistant Mycoplasma pneumoniae in humans, Ontario, Canada, 2010–2011". Emerg Infect Dis. 19. doi:10.3201/eid1909.121466.
- 1 2 3 Meyers, L. A.; Newman, M. E. J.; Martin, M.; Schrag, S. "Applying Network Theory to Epidemics: Control Measures for Mycoplasma pneumoniae Outbreaks". Emerg. Infect. Dis. 9: 204–210. doi:10.3201/eid0902.020188.
This article incorporates public domain text from the CDC as cited.
Further reading
- Baseman J. B. (1996). "Interplay between Mycoplasma Surface Proteins, Airway Cells, and the Protean Manifestations of Mycoplasma-mediated Human Infections". American Journal of Respiratory and Critical Care Medicine. 154: 137–144. doi:10.1164/ajrccm/154.4_Pt_2.S137.
- Razin S.; Yogev D.; Naot Y. (1998). "Molecular biology and pathogenicity of mycoplasmas". Microbiol. Mol. Biol. Rev. 62 (4): 1094–156. PMC 98941. PMID 9841667.
- Kashyap S., Sarkar, M. (2010). "Mycoplasma pneumonia: Clinical features and management". Lung India. 27 (2): 75–85. doi:10.4103/0970-2113.63611.
- Narita M. (2009). "Pathogenesis of neurologic manifestations of Mycoplasma pneumoniae infection". Pediatr Neurol. 41 (3): 159–166. doi:10.1016/j.pediatrneurol.2009.04.012. PMID 19664529.
- Ferwerda A.; Moll H. A.; de Groot R. (2001). "Respiratory tract infections by Mycoplasma pneumoniae in children: a review of diagnostic and therapeutic measures". Eur J Pediatr. 160 (8): 483–491. doi:10.1007/s004310100775. PMID 11548186.
- Esposito S.; Droghetti R.; Bosis S.; Claut L. Marchisio P.; Principi N. (2002). "Cytokine secretion in children with acute Mycoplasma pneumoniae infection and wheeze". Pediatric Pulmonology. 34 (2): 122–127. doi:10.1002/ppul.10139.
- Ríos A. M.; Mejías A.; Chávez-Bueno S.; Fonseca-Aten M.; Katz K.; Hatfield J.; Gómez A. M.; Jafri H. S.; McCracken G. H.; Ramilo O.; Hardy R. D. (2004). "Impact of Cethromycin (ABT-773) Therapy on Microbiological, Histologic, Immunologic, and Respiratory Indices in a Murine Model of Mycoplasma pneumoniae Lower Respiratory Infection". Antimicrob. Agents Chemother. 48 (8): 2897–2904. doi:10.1128/AAC.48.8.2897-2904.2004.
External links
- Mycoplasma pneumoniae genome
- Type strain of Mycoplasma pneumoniae at BacDive - the Bacterial Diversity Metadatabase