Multipolar exchange interaction
Magnetic materials with strong spin-orbit interaction, such as: LaFeAsO,[1] PrFe4P12[2] , YbRu2Ge2,[3] UO2,[4] NpO2,[5] Ce1−xLaxB6,[6] URu2Si2[7] and many other compounds, are found to have magnetic ordering constituted by high rank multipoles, e.g. quadruple, octople, etc.[8] Due to the strong spin-orbit coupling, multipoles are automatically introduced to the systems when the total angular momentum quantum number J is larger than 1/2. If those multipoles are coupled by some exchange mechanisms, those multipoles could tend to have some ordering as conventional spin 1/2 Heisenberg problem. Except the multipolar ordering, many hidden order phenomena are believed closely related to the multipolar interactions [5][6][7]
Tensor Operators Expansion
Basic Concepts
Consider a quantum mechanical system with Hilbert space spanned by , where is the total angular momentum and is its projection on the quantization axis. Then any quantum operators can be represented using the basis set as a matrix with dimension . Therefore, one can define matrices to completely expand any quantum operator in this Hilbert space. Taking J=1/2 as an example, a quantum operator A can be expanded as







Obviously, the matrices: forms a basis set in the operator space. Any quantum operator defined in this Hilbert can be expended by operators. In the following, let's call these matrices as a super basis to distinguish the eigen basis of quantum states. More speifically the above super basis can be called transition super basis because it describes the transition between states and . In fact, this is not the only super basis that does the trick. We can also use Pauli matrices plus identity matrix to form a super basis





Since the rotation properties of follow the same rules as the rank 1 tensor of cubic harmonics and the identity matrix follows the same rules as the rank 0 tensor , the basis set can be called cubic super basis. Another commonly used super basis is spherical harmonic super basis which is built by replacing the to the raising and lowering operators








Again, share the same rotational properties as rank 1 spherical harmonic tensors , so it is called spherical super basis.


Because atomic orbitals are also described by spherical or cubic harmonic functions, one can imagine or visualize these operators using the wave functions of atomic orbitals although they are essentially matrices not spatial functions.

If we extend the problem to , we will need 9 matrices to form a super basis. For transition super basis, we have . For cubic super basis, we have . For spherical super basis, we have . In group theory, are called scalar or rank 0 tensor, are called dipole or rank 1 tensors, are called quadrupole or rank 2 tensors.[8]







The example tells us, for a -multiplet problem, one will need all rank tensor operators to form a complete super basis. Therefore, for a system, its density matrix must have quadrupole components. This is the reason why a problem will automatically introduce high-rank multipoles to the system [9]



Formal Definitions

A general definition of spherical harmonic super basis of a -multiplet problem can be expressed as [8]

where the parentheses denote a 3-j symbol; K is the rank which ranges ; Q is the projection index of rank K which ranges from −K to +K. A cubic harmonic super basis where all the tensor operators are hermitian can be defined as
![T_{K}^{Q} =\frac{1}{\sqrt{2}}[(-1)^{Q}Y_{K}^{Q}(J)+Y_{K}^{-Q}(J)]](../I/m/4dcfd05c388393bbed7b31c8d552ffedd5786f23.svg)
![T_{K}^{-Q} =\frac{i}{\sqrt{2}}[Y_{K}^{-Q}(J)-(-1)^{Q}Y_{K}^{-Q}(J)]](../I/m/d566df15250fd41f0c3fa593a0616f64f4bff7cf.svg)
Then, any quantum operator defined in the -multiplet Hilbert space can be expanded as


where the expansion coefficients can be obtained by taking the trace inner product, e.g. . Apparently, one can make linear combination of these operators to form a new super basis that have different symmetries.
![\alpha_{K}^{Q}=Tr[AY_{K}^{Q\dagger}]](../I/m/8807b24804e7dbf6355468abcae449650791e2f7.svg)
Multi-exchange Description
Using the addition theorem of tensor operators, the product of a rank n tensor and a rank m tensor can generate a new tensor with rank n+m ~ |n-m|. Therefore, a high rank tensor can be expressed as the product of low rank tensors. This convention is useful to interpret the high rank multipolar exchange terms as a "multi-exchange" process of dipoles (or pseudospins). For example, for the spherical harmonic tensor operators of case, we have





If so, a quadrupole-quadrupole interaction (see next section) can be considered as a two steps dipole-dipole interaction. For example, , so the one step quadrupole transition on site now becomes a two steps of dipole transition . Hence not only inter-site-exchange but also intra-site-exchange terms appear (so called multi-exchange). If is even larger, one can expect more complicated intra-site-exchange terms would appear. However, one has to note that it is not a perturbation expansion but just a mathematical technique. The high rank terms are not necessarily smaller than low rank terms. In many systems, high rank terms are more important than low rank terms.[8]




Multipolar Exchange Interactions


There are four major mechanisms to induce exchange interactions between two magnetic moments in a system:[8] 1). Direct exchange 2). RKKY 3). Superexchange 4). Spin-Lattice. No matter which one is dominated, a general form of the exchange interaction can be written as[9]

where are the site indexes and is the coupling constant that couples two multipole moments and . One can immediately find if is restricted to 1 only, the Hamiltonian reduces to conventional Heisenberg model.





An important feature of the multipolar exchange Hamiltonian is its anisotropy.[9] The value of coupling constant is usually very sensitive to the relative angle between two multipoles. Unlike conventional spin only exchange Hamiltonian where the coupling constants are isotropic in a homogeneous system, the highly anisotropic atomic orbitals (recall the shape of the wave functions) coupling to the system's magnetic moments will inevitably introduce huge anisotropy even in a homogeneous system. This is one of the main reasons that most multipolar orderings tend to be non-colinear.
Antiferromagnetism of Multipolar Moments

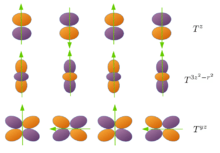
Unlike magnetic spin ordering where the antiferromagnetism can be defined by flipping the magnetization axis of two neighbor sites from a ferromagnetic configuration, flipping of the magnetization axis of a multipole is usually meaningless. Taking a moment as an example, if one flips the z-axis by making a rotation toward the y-axis, it just changes nothing. Therefore, a suggested definition[9] of antiferromagnetic multipolar ordering is to flip their phases by , i.e. . In this regard, the antiferromagnetic spin ordering is just a special case of this definition, i.e. flipping the phase of a dipole moment is equivalent to flipping its magnetization axis. As for high rank multipoles, e.g. , it actually becomes a rotation and for it is even not any kind of rotation.




Compute Coupling Constants
Calculation of multipolar exchange interactions remains a challenging issue in many aspects. Although there were many works based on fitting the model Hamiltonians with experiments, predictions of the coupling constants based on first-principle schemes remain lacking. Currently there are two studies implemented first-principles approach to explore multipolar exchange interactions. An early study was developed in 80's. It is based on a mean field approach that can greatly reduce the complexity of coupling constants induced by RKKY mechanism, so the multipolar exchange Hamiltonian can be described by just a few unknown parameters and can be obtained by fitting with experiment data.[10] Later on, a first-principles approach to estimate the unknown parameters was further developed and got good agreements with a few selected compounds, e.g. cerium momnpnictides.[11] Another first-principle approach was also proposed recently.[9] It maps all the coupling constants induced by all static exchange mechanisms to a series of DFT+U total energy calculations and got agreement with uranium dioxide.
References
- ↑ F. Cricchio, O. Granas, and L. Nordstrom, Phys. Rev. B. 81, 140403 (2010); R. S. Gonnelli, D. Daghero, M. Tortello, G. A. Ummarino, V. A. Stepanov, J. S. Kim, and R. K. Kremer, Phys. Rev. B 79, 184526 (2009)
- ↑ A. Kiss and Y. Kuramoto, J. Phys. Soc. Jpn. 74, 2530 (2005); H. Sato, T. Sakakibara, T. Tayama, T. Onimaru, H. Sugawara, and H. Sato, J. Phys. Soc. Jpn. 76, 064701 (2007)
- ↑ T. Takimoto and P. Thalmeier, Phys. Rev. B 77, 045105 (2008)
- ↑ S.-T. Pi, R. Nanguneri, and S. Savrasov, Phys. Rev. Lett. 112, 077203 (2014); P. Giannozzi and P. Erdos, J. Mag. Mag Mater. 67, 75 (1987). V. S. Mironov, L. F. Chibotaru, and A. Ceulemans, Adv. Quan. Chem. 44, 599 (2003); S. Carretta, P. Santini, R. Caciuffo, and G. Amoretti, Phys. Rev. Lett. 105, 167201 (2010); R. Caciuffo, P. Santini, S. Carretta, G. Amoretti, A. Hiess, N. Magnani, L. P. Regnault, and G. H. Lander, Phys. Rev. B 84, 104409 (2011)
- 1 2 P. Santini and G. Amoretti, Phys. Rev. Lett 85, 2188 (2000); P. Santini, S. Carretta, N. Magnani, G. Amoretti, and R. Caciuffo, Phys. Rev. Lett. 97, 207203 (2006); K. Kubo and T. Hotta, Phys. Rev. B 71, 140404 (2005)
- 1 2 D. Mannix, Y. Tanaka, D. Carbone, N. Bernhoeft, and S. Kunii, Phys. Rev. Lett. 95, 117206 (2005)
- 1 2 P. Chandra, P. Coleman, J. A. Mydosh, and V. Tripathi, Nature (London) 417, 831 (2002); Francesco Cricchio, Fredrik Bultmark, Oscar Granas, and Lars Nordstrom, Phys. Rev. Lett. 103, 107202 (2009); Hiroaki Ikeda, Michi-To Suzuki, Ryotaro Arita, Tetsuya Takimoto, Takasada Shibauchi, and Yuji Matsuda, Nat. Phys. 8, 528 (2012); A. Kiss and P. Fazekas, Phys. Rev. B 71, 054415 (2005); J. G. Rau and H.-Y. Kee, Phys. Rev. B 85, 245112 (2012)
- 1 2 3 4 5 R. Caciuffo et al., Rev. Mod. Phys. 81, 807 (2009)
- 1 2 3 4 5 6 7 8 9 S.-T. Pi, R. Nanguneri, and S. Savrasov, Phys. Rev. Lett 112, 077203 (2014); S.-T. Pi, R. Nanguneri, and S. Savrasov, Phys. Rev. B 90, 045148 (2014)
- ↑ R. Siemann and B. R. Cooper, Phys. Rev. Lett. 44, 1015 (1980)
- ↑ J. M. Wills and B. R. Cooper, Phys. Rev. B 42, 4682 (1990)