Kallmann syndrome
| Kallmann syndrome | |
|---|---|
 | |
|
The structure of GNRH1 (from PDB: 1YY1) | |
| Classification and external resources | |
| Specialty | endocrinology |
| ICD-10 | E23.0 |
| ICD-9-CM | 253.4 |
| OMIM | 308700 147950 244200 138850 607002 146110 136350 615271 615270 614880 1527600 162330 164160 608137 608892 300473 603286 613301 604808 603725 606807 602748607984 |
| DiseasesDB | 7091 |
| eMedicine | med/1216 med/1342 |
| MeSH | D017436 |
Kallmann syndrome is a rare genetic condition that is characterized by a failure to start or a failure to complete puberty. It is also accompanied by a lack of sense of smell (anosmia) or a highly reduced sense of smell (hyposmia). The condition can occur in both males and females but is more commonly diagnosed in males. Left untreated, patients with Kallmann syndrome will almost invariably be infertile.[1][2]
Kallmann syndrome occurs due to a failure of the hypothalamus to release GnRH at the appropriate time as a result of the GnRH releasing neurones not migrating into the correct location during embryonic development.[3][4]
Kallmann syndrome is a form of hypogonadotropic hypogonadism (HH). Approximately 50% of HH cases occur with no sense of smell and are classified as Kallmann syndrome. Apart from the sense of smell there is no difference in the diagnosis or treatment between a case of HH or a case of Kallmann syndrome.[5][6]
The terminology used when describing cases of HH can vary. The term congenital hypogonadotropic hypogonadism (CHH) is now often used. Other terms used include idiopathic / isolated hypogonadotropic hypogonadism (IHH), normosmic hypogonadotropic hypogonadism (nHH) or hypothalamic hypogonadism. The term HH can be used to cover all cases, including Kallmann syndrome.
The term isolated GnRH deficiency (IGD) has increasingly been used to describe this group of conditions as it highlights the primary cause of these conditions and distinguishes them from other conditions such as Klinefelter syndrome or Turner syndrome which share some similar symptoms but have a totally different etiology.[7]
The term hypogonadism describes a low level of circulating sex hormones; testosterone in males and estrogen and progesterone in females. Hypogonadism can occur through a number of different methods. The use of the term hypogonadotropic relates to the fact that the hypogonadism found in HH is caused by a disruption in the production of the gonadotropin hormones normally released by the anterior pituitary gland known as luteinising hormone (LH) and follicle stimulating hormone (FSH).
LH and FSH have a direct action on the ovaries in women and testes in men. The absence of LH and FSH means that initially puberty will not commence at the correct time and subsequently the ovaries and testes will not perform their normal fertility function with the maturation and release of eggs in women and the production of sperm in men alongside their role in producing the sex hormones.
The underlying cause of the failure in production of LH and FSH is the impairment of the hypothalamus to release the hormone GnRH which in normal circumstances induces the production of LH and FSH. Without the correct release of GnRH the pituitary gland is unable to release LH and FSH which in turn prevents the ovaries and testes from functioning correctly. This failure in GnRH production can either be due to the absence of the GnRH releasing neurones inside the hypothalamus or the inability of the hypothalamus to release GnRH in the correct pulsatile manner to ensure LH and FSH release from the pituitary.[8]
HH can occur as an isolated condition with just the LH and FSH production being affected or it can occur in combined pituitary deficiency conditions such as CHARGE syndrome.
To date at least twenty five different genes have so far been implicated in causing Kallmann syndrome or other forms of HH through a disruption in the production or activity of GnRH. The genes involved cover all forms of inheritance and no one gene defect has been shown to be common to all cases which makes genetic testing and inheritance prediction very problematic.[9]
Kallmann syndrome was described in a paper published in 1944 by Franz Josef Kallmann, a German-American geneticist.[10][11] The link between anosmia and hypogonadism had already been noted however, in particular by the Spanish doctor Aureliano Maestre de San Juan in 1856.[12] The condition is sometimes known by his name in Spanish speaking countries.
The condition has a low prevalence, estimated as being between 1 in 4,000 and 1 in 10,000 for male HH cases overall and 1 in 50,000 for Kallmann syndrome. It is three to five times more common in males than females. Though whether this is a true sex imbalance or a reflection on how difficult KS / HH is to diagnose correctly, especially in females, has yet to be fully established.[13][14][15] A more recent paper published in 2011[16] gave the incidence in the Finnish population at 1 in 48,000, with a sex distinction of 1 in 30,000 for males and 1 in 125,000 for females.
Signs and symptoms
The features of Kallmann syndrome (KS) and hypogonadotropic hypogonadism (HH) can be split into two different categories; "reproductive" and "non reproductive". Not all symptoms will appear in every case of KS/HH, not even amongst family members. Some of these features are linked to the gene defects known to cause KS/HH but in some cases it is still not clear why some of these features exist. It has been estimated that 60% of KS/HH cases will show a non-reproductive symptom.
It is normally difficult to distinguish a case of KS/HH from a straightforward constitutional delay of puberty. However, if puberty has not commenced by either 14 (girls) or 15 (boys) and one of these non-reproductive features are present then a referral to reproductive endocrinologist might be advisable.
Kallmann syndrome and hypogonadotropic hypogonadism do not exist as distinct conditions. Each case can show a different range of symptoms and a different severity of symptoms. Even family members will not always show the same degree of symptoms. Cases of KS/HH can be separated into different categories depending on the gene mutation(s) involved.[1][8] Severity can range from total absence of puberty with anosmia to slightly delayed puberty with or without anosmia.
- Classic HH
This type of HH is present from birth and is lifelong. Approximately two-thirds of Classic HH cases will have a low level of pulsatile GnRH release from the hypothalamus which will give rise to partial puberty while the other third of cases will have zero GnRH release and no puberty.
The non-reproductive symptoms mentioned earlier in this article will be present in approximately half the cases. The most common of these is anosmia, which gives rise to the distinction between KS and HH. Males with classic HH may also have had a history of un-descended testicles and/or micropenis.
This type of HH has been shown to be caused by polyallelic mutations in males and females and autosomal dominant and x-linked recessive mutations in males mentioned in the table earlier in this article.
- Adult-onset HH
This type of HH has only been shown to occur in males. The hypothalamic-pituitary-gonadal axis (HPG axis) functions normally at birth and well into adult life giving normal puberty.
The HPG axis then either fails totally or is reduced to a very low level of GnRH release, in adult life with no obvious cause such as a pituitary tumor. This will lead to a fall in testosterone levels and infertility. This type of HH is not associated with any non-reproductive symptoms, and it has been shown to be caused by monoallelic mutations.
This type of KS/HH will appear to be the classic lifelong form at first but at some point in adult life the HPG axis resumes its normal function and GnRH, LH, and FSH levels return to normal levels. Has only been shown to occur in 10% of cases, primarily KS cases rather than HH cases and only found in patients who have undergone some form of testosterone replacement therapy. It is only normally discovered when testicular volume increases while on testosterone treatment alone and testosterone levels return to normal when treatment is stopped.
This type of KS/HH rarely occurs in cases where males have had a history of un-descended testes and/or micropenis and has been shown to be caused by monoallelic mutations.
- Hypothalamic amenorrhoea[19]
This type of HH is seen in females where the HPG axis is suppressed in response to physical or psychological stress or malnutrition. It is reversible with the removal of the stressor.
This type of HH is not associated with any non-reproductive symptoms and has been shown to be caused by monoallelic mutations. A study suggested that there may have been an evolutionary advantage at one stage in the early development of man for these genes to exist where it could have been an advantage for the females not to be fertile at times when food was scarce in the community.
This type of HH has been shown to be caused by monoallelic mutations.
Reproductive features
- Failure to start or fully complete puberty in both men and women
- Lack of testicle development in men; size < 3 cm3. The normal range is between 12 and 30 cm3
- Primary amenorrhoea or failure to start menstruation in women
- Poorly defined secondary sexual characteristics in both men and women.
- Infertility
Non-reproductive features[5][20]
- Hypogonadotropic hypogonadism (a lack of the pituitary hormones luteinizing hormone and follicle-stimulating hormone)
- Congenital (present from birth)
- Total lack of sense of smell (anosmia) or markedly reduced sense of smell (hyposmia). This is the defining feature of Kallmann syndrome; it is not seen in other cases of HH. Approximately 50% of HH cases occur with anosmia and can be termed as Kallmann syndrome.
- Cleft palate or other craniofacial defects.
- Skeletal defects including scoliosis.[21]
- Unilateral renal agenesis or aplasia; absence or non-functioning of one of the kidneys
- Cryptorchidism, un-descended testicles at birth, occurs in 30% of KS/HH cases
- Micropenis, occurs in fewer than 5 to 10% of KS/HH cases.
- Neural hearing defects
- Synkinesis or mirror movements of hands
- Dental defects
- Normal stature, but there can be an increase in height if treatment is delayed, due to the lack of testosterone or oestrogen causing excess bone growth in the arms and legs.
At one stage it was thought that colour blindness was linked to KS/HH but this has proved not to be the case.
A fraction of cases may present with post-pubertal onset, which results in a phenotypically normal penis in men with subsequent testicular atrophy and loss of some secondary sex traits. These men generally present with sexual impairment and low libido. In women, late-onset Kallmann Syndrome can result in secondary amenorrhoea. Anosmia may or may not be present in these individuals.
Patients with KS and other forms of HH are almost invariably born with normal sexual differentiation; i.e., they are physically male or female. This is due to the human chorionic gonadotrophin (hCG) produced by placenta at approximately 12 to 20 weeks gestation (pregnancy) which is normally unaffected by having KS or HH.
Patients with KS/HH lack a surge of GnRH, LH, and FSH that occurs between birth and six months of age.[22] This surge is particularly important in infant boys as it helps with testicular descent into the scrotum. A small percentage of boys with KS/HH will be born with micropenis and/or undescended testes, both of which may be treated surgically in the first year of life. The surge of GnRH/LH/FSH in non KS/HH children gives detectable levels of testosterone in boys and oestrogen & progesterone in girls. The lack of this surge can sometimes be used as a diagnostic tool if KS/HH is suspected in a newborn boy, but is not distinct enough for diagnosis in girls.
Osteoporosis
One possible side effect of having KS/HH is the increased risk of developing secondary osteoporosis or osteopenia. Oestrogen (females) or testosterone (males) is essential for maintaining bone density.[6] Deficiency in either testosterone or oestrogen can increase the rate of bone resorption while at the same time slowing down the rate of bone formation. Overall this can lead to weakened, fragile bones which have a higher tendency to fracture.
Even a short time with low oestrogen or testosterone, as in cases of delayed diagnosis of KS/HH can lead to an increased risk of developing osteoporosis but other risk factors are involved so the risk of developing it will vary from patient to patient.
Patients with KS/HH should have a bone density scan at least every five years, even if they are on constant hormone replacement therapy. This interval will be shortened to three years if the patient is already in the at-risk zone (osteopenia) or yearly if the patient has osteoporosis already.
The bone density scan is known as a dual energy X-ray absorptiometry scan (DEXA or DXA scan). It is a very simple straightforward test, taking less than 15 minutes to perform. It involves taking a specialised X-ray picture of the spine and hips and measuring the bone mineral density and comparing the result to the average value for a young healthy adult in the general population.[23]
Adequate calcium levels, and probably more importantly Vitamin D levels are essential for healthy bone density. Some patients with KS/HH will have their levels checked and may be prescribed extra Vitamin D tablets or injections to try to prevent the condition getting worse. The role of Vitamin D for general overall health is under close scrutiny at the moment with some researchers claiming Vitamin D deficiency is prevalent in many populations and can be linked to other disease states.
Some people with severe osteoporosis might be prescribed bisphosphonates. Exercise, especially weight bearing and resistance exercise, is known to reduce the risk of osteoporosis.
Reversal of symptoms
Reversal of symptoms have been reported in between 15% to 22% of cases.[24] The causes of this reversal are still under investigation but have been reported in both males and females.
Reversal appears to be associated with 14 of the known gene defects linked to KS/CHH. The study suggests no obvious gene defect showing a tendency to allow reversal. There is a suggestion that the TAC3 and TACR3 mutations might allow for a slightly higher chance of reversal but the numbers involved are too low to confirm this. The KAL-1 mutation appears to be least likely to allow reversal with to date only one recorded instance in medical literature. Even male patients who previous had micro-phallus or cryptorchidism have been shown to undergo reversal of symptoms.
The reversal might not be permanent and remission can occur at any stage; the paper suggests that this could be linked to stress levels. The paper highlighted a reversal case that went into remission but subsequently achieved reversal again, strongly suggesting an environmental link.
Reversal cases have been seen in cases of both Kallmann syndrome and congenital hypogonadotropic hypogonadism but appear to be less common in cases of Kallmann syndrome where the sense of smell is also affected. A paper published in 2016[25] agreed with the theory that there is a strong environmental or epigenetic link to the reversal cases. The precise mechanism of reversal is unclear and is an area of active research.
Reversal would be apparent if testicular development was seen in men while on testosterone therapy alone or in women who menstruate or achieved pregnancy while on no treatment. To date there have been no recorded cases of the reversal of anosmia found in Kallmann syndrome cases.
Diagnosis
The diagnosis is often one of exclusion found during the workup of delayed puberty.[14][26][27]
A paper published in 2012 by Prof. Jacques Young[28] highlights a typical example of the diagnostic work up involved in a suspected case of KS/HH.
One of the biggest problems in the diagnosis of Kallmann syndrome and other forms of HH is the ability to distinguish between a normal constitutional delay of puberty and Kallmann syndrome (KS) or hypogonadotropic hypogonadism (HH).[29]
The main biochemical parameters in men are low serum testosterone and low levels of the gonadotropins LH and FSH, and in women low serum oestrogen and low levels of LH and FSH.
For both males and females with constitutional delay of puberty, endogenous puberty will eventually commence without treatment. However a delay in treatment in a case of KS/HH will delay the physical development of the patient and can cause severe psychological damage. The "wait and see" approach applied to "late bloomers" is probably counterproductive to the needs of the patient whereas a step-by-step approach with hormone replacement therapy used with slowly increasing doses can be used as a diagnostic tool.
Normally testicular enlargement is the key sign for the onset of puberty in boys however the use of nighttime LH sampling can help predict the onset of puberty.
In females diagnosis is sometimes further delayed as other causes of amenorrhoea normally have to be investigated first before a case of KS/HH is considered.[30] Normally the onset of puberty is marked in females by the onset of menstruation however KS / HH can still occur in females in cases when menstruation has begun but stopped after one or two menstrual bleeds. A study of GnRH deficient women in 2011[31] showed that 10% had experienced one or two bleeds before the onset of amenorrhoea.
In males, treatment with age-appropriate levels of testosterone can be used to distinguish between a case of KS/HH from a case of delayed puberty. If just delayed the testosterone can "kick-start" endogenous puberty, as demonstrated by testicular enlargement, whereas in the case of KS/HH there will be no testicular enlargement while on testosterone therapy alone. If no puberty is apparent, especially no testicular development, then a review by a reproductive endocrinologist may be appropriate. Dr Richard Quinton, a leading UK expert on KS/HH, suggests that if puberty is not apparent by the age of 16 then the patient should be referred for endocrinological review.[32]
A full endocrine workup will be required to measure the levels of the other pituitary hormones, especially prolactin, to check that the pituitary gland is working correctly. There can be other general health issues such as being overweight or having an underlying chronic or acute illness which could cause a delay of puberty. This makes it essential for a patient to get a full endocrine review to distinguish between a case of KS/HH and another cause for the pubertal delay.
Bone age can be assessed using hand and wrist X-rays. If the bone age is significantly lower than the chronological age of the patient, this could suggest delayed puberty unless there is another underlying reason for the discrepancy.
A karyotype may be performed to rule out Klinefelter syndrome and Turner syndrome, although the hormones levels would also rule out both these relatively common reasons for hypogonadism.
A magnetic resonance imaging (MRI) scan can be used to determine whether the olfactory bulb is present and to check for any physical irregularities of the pituitary gland or hypothalamus.
A standard smell test can be used to check for anosmia, but it must be remembered that even in total anosmia various substances (such as menthol and alcohol) can still be detected by direct stimulation of the trigeminal nerve.
Genetic screening can be carried out, especially for the KAL-1 mutation, but in light of the uncertain genetic origin of the majority of KS and HH cases a negative result will not rule out a possible diagnosis.
A review paper published in 2014[33] highlighted the need for doctors to be aware of the possible diagnosis of KS / HH if pubertal delay is found alongside associated "red flag" symptoms. The symptoms listed in the paper were split into two categories; reproductive symptoms associated with the lack of mini puberty seen between birth and six months of age and non-reproductive symptoms which are associated with specific forms of HH. As with other review papers the authors also warned against the "wait and see" approach when puberty appears to be delayed.
Reproductive features:
Non-reproductive features:
deafness (normally nerve deafness)
synkinesis of the hands (mirror movements)
renal agenesis (absence of one the kidneys)
dental anomalies
syndactyly or other anomalies of the hands
Pathophysiology
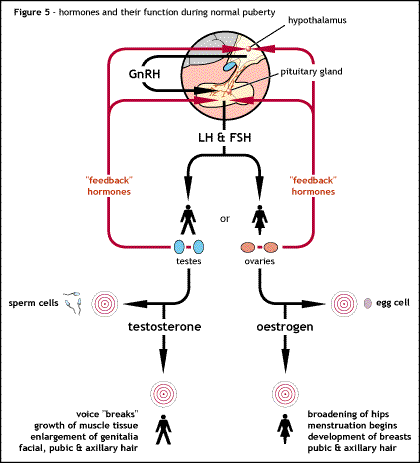
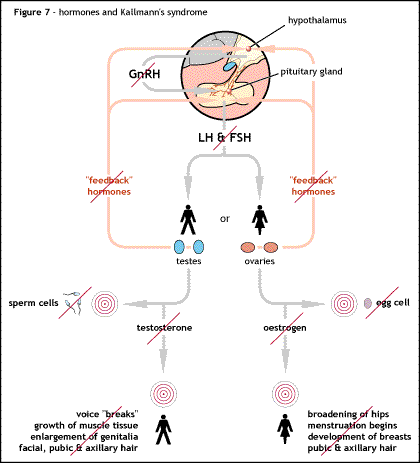
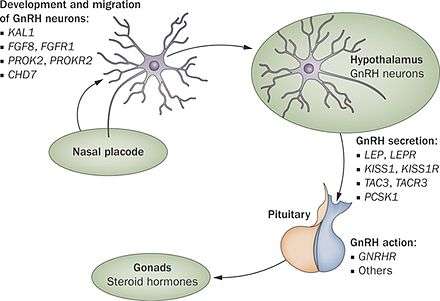
Kallmann syndrome (KS) and other forms of hypogonadotropic hypogonadism (HH) are classed as pituitary or endocrine disorders. While the end result is a failure of puberty and the development of secondary sexual characteristics, the underlying cause of the disorder is located between the two endocrine glands located within the brain.
The hypothalamus gland and the pituitary gland can be seen as the control stations for all the hormonal activity throughout the body. These glands secrete a number of different hormones with various effects around the body. KS/HH results from the disruption in the communication between the hypothalamus and pituitary in regard to one set of hormones only. All the other actions of the hypothalamus and pituitary glands remain unaffected.
Normally the hypothalamus releases a hormone called gonadotropin releasing hormone (GnRH). GnRH is released from the hypothalamus in a pulsatile manner at set intervals throughout the day via the hypophyseal portal system and acts on the anterior pituitary gland, causing it to release two hormones called gonadotropins. These hormones are luteinising hormone (LH) and follicle stimulating hormone (FSH), which have a direct action on the testes in men and ovaries in women. LH and FSH are essential for stimulating the development of secondary sexual characteristics seen at puberty and for maintaining the normal sexual function of both men and women, including maintaining the correct levels of the sex steroids: testosterone in men and oestrogen and progesterone in women.
In KS/HH the release of GnRH is either totally blocked or vastly reduced. The GnRH is released by the hypothalamus by specialised nerve cells or neurones. During development of the brain in the first 10 weeks of development these GnRH releasing neurones migrate from their original source and end up inside the hypothalamus.
The GnRH neurones originate in an area of the developing brain called the olfactory placode; they then pass through the cribriform plate and into a structure called the olfactory bulb, where the sense of smell is generated. From there they migrate into what will be become the hypothalamus. Any problems with the development of the olfactory bulb will prevent the progression of the GnRH releasing neurones through it. If the GnRH releasing neurones are prevented from reaching the hypothalamus no GnRH will be released, so in turn no FSH or LH will be released which results in the failure of puberty and deficient production of testosterone in men, and oestrogen and progesterone in women.
In Kallmann syndrome the olfactory bulb is missing or not fully developed which gives rise to the additional symptom of lack of sense of smell (anosmia) or vastly reduced sense of smell (hyposmia). In other forms of HH the olfactory bulb develops correctly, so there is a normal sense of smell but the migration of the GnRH releasing neurones is affected elsewhere and still prevents GnRH being released at the correct time.
However, this connection between the olfactory bulb and GnRH neuron cells is not as tightly coupled as first thought. The majority of congenital anosmics with no olfactory bulbs do not have reproductive hormone deficiencies.[34] In the same family with the same KS/HH gene(s), some members may have KS with no sense of smell, others may have IHH with a sense of smell and others may have isolated anosmia and no hormone deficiencies.
The majority of the genes that have been associated with causing cases of KS or HH play a part in either the generation, migration, or activity of these GnRH releasing neurones and their ability to stimulate FSH and LH production.
| OMIM | Name | Gene | Locus | Description |
|---|---|---|---|---|
| 308700 | KAL1 | KAL1 | Xp22.3 | Kallmann syndrome can be inherited as an X-linked recessive trait, in which case there is a defect in the KAL1 gene, which maps to chromosome Xp22.3.[2] KAL encodes a neural cell adhesion molecule, anosmin-1. Anosmin-1 is normally expressed in the brain, facial mesenchyme, mesonephros and metanephros. It is required to promote migration of GnRH neurons into the hypothalamus. It also allows migration of olfactory neurons from the olfactory bulbs to the hypothalamus. |
| 147950 | KAL2 | FGFR1 | 8p11.2-p11.1 | An autosomal dominant gene on chromosome 8 {8p12} (KAL-2 or FGFR-1 (fibroblast growth factor receptor 1)) is thought to cause about 10% of cases. |
| 244200 | KAL3 | PROKR2 | 20p13 | An additional autosomal cause of Kallmann syndrome has been reported[35] to be mutations in the prokineticin receptor-2 gene (PROKR2)(KAL-3) at position 20p13 and its ligand prokineticin 2 (PROK2)(KAL-4) at position 3p21.1. It was noted that mutations in these genes brought about various degrees of olfactory and reproductive dysfunction, but not the other symptoms seen in the KAL-1 and KAL-2 forms of Kallmann Syndrome. The authors of the paper suggested that up to 30% of all Kallmann Syndrome cases can be linked to known genetic mutations. |
| 1527600 | GnRH1 | GnRH1 | 8p21-11.2 | Ligand for the GnRH receptor GnRH11527600 at locus 4q21.2. Binding allows FSH/LH secretion by the pituitary gland. Known to cause IHH and partial IHH. |
| 162330 | TAC3 | TAC3 | 12q13-21 | Ligand for the TAC receptor TACR3 162332 at locus 4q25. Signalling peptide, crucial for GnRH secretion. Known to cause severe IHH with high incidence of micropenis. Known to be one of the genes that have shown a higher rate of reversible IHH than other genes. |
| 164160 | LEP | LEP | 7q31.2 | Ligand for the receptor LRPR. Involved in pulsatile GnRH secretion. |
| 600483 | FGF8 | FGF8 | 10q24.32 | Fibroblast growth factor 8.600483 Associated with cleft lip and / or palate and hearing loss.[36] |
| 608137 | NELF | NELF | 9q34.3 | Not associated with ligand/receptor binding. Associated with the migration of the olfactory and GnRH neurones during development. |
| 608892 | CHD7 | CHD7 | 8q12.1 | Not associated with ligand/receptor binding. Associated with original generation of GnRH and olfactory neurones. Strongly associated with CHARGE syndrome. |
| 300473 | DAX1/NROB1 | DAX1 | Xp21.3-21.2 | Nuclear receptor with no known ligand. Known to be a transcription inhibitor. Thought to cause X-linked recessive forms of IHH in both males and occasionally females. Known to cause pubertal delay in females. |
| 603286 | KiSS1 | KiSS-1 | 1q32.1 | Ligand for receptor KiSS1R 604161 at locus 19p13.3. Peptide produced by the hypothalamus, essential for pulsatile GnRH secretion. Known to cause IHH, thought to be involved in the timing of the onset of puberty. |
| 613301 | FEZF1 | FEZF1 | 7q31.32 | Protein modification (zinc finger) which facilitates the movement of the olfactory neurones in the early development of the brain. A mutation in this protein has be shown to prevent the passage of GnRH releasing neurones into the hypothalamus. A 2014 paper highlighted this gene in causing HH in two families.[37] |
| 604808 | FLRT3 | FLRT3 | 20p12.1 | Fibronectin-like domain-containing leucine rich transmembrane protein 3. Protein associated with the function of the KAL-2 gene (fibroblast growth factor and its related receptor) which allows for the migration of both olfactory and GnRH releasing neurones during early embryonic development.[38] |
| 603725 | FGF17 | FGF17 | 8p21.3 | Fibroblast growth factor 17. Protein associated with the function of the KAL-2 gene (fibroblast growth factor and its related receptor) which allows for the migration of both olfactory and GnRH releasing neurones during early embryonic development.[38] |
| 606807 | IL17RD | IL17RD | 3p14.3 | Interleukin receptor 17 D. Protein associated with the function of the KAL-2 gene (fibroblast growth factor and its related receptor) which allows for the migration of both olfactory and GnRH releasing neurones during early embryonic development.[38] |
| 602748 | DUSP6 | DUSP6 | 12q21.33 | Dual specificity phosptate-6. Protein associated with the function of the KAL-2 gene (fibroblast growth factor and its related receptor) which allows for the migration of both olfactory and GnRH releasing neurones during early embryonic development.[38] |
| 607984 | SPRY4 | SPRY4 | 5q21.3 | Sprouty, Drosphila, homolog of, 4. Protein associated with the function of the KAL-2 gene (fibroblast growth factor and its related receptor) which allows for the migration of both olfactory and GnRH releasing neurones during early embryonic development.[38] |
A 2007 paper proposed a possible digenic model for Kallmann syndrome and other forms of hypogonadotrophic hypogonadism.[1] The possibility of two separate gene defects working in combination could account for some of the variation of symptoms seen in cases of Kallmann syndrome, even within families.
The genetics of Kallmann syndrome and other forms of hypogonadotrophic hypogonadism is still far from clear with around 70% of cases having an unknown genetic origin.[39]
Further research published by Anna Mitchell et al.[8] has highlighted the fact that the number of genetic loci known to cause cases of Kallmann syndrome and HH has increased to ten. The paper highlights the broad spectrum of physical symptoms—both reproductive and non-reproductive—that can occur in cases of Kallmann syndrome and HH, even within the same family group.
Treatment
Treatment for KS and other forms of HH can be divided into hormone replacement therapy and fertility treatments.[22][26][27][40]
Hormone replacement therapy
The aim for hormone replacement therapy (HRT) for both men and women is to ensure that the level of circulating hormones (testosterone for men and oestrogen/progesterone for women) is at the normal physiological level for the age of the patient. At first the treatment will produce most of the physical and psychological changes seen at puberty, with the major exception that there will be no testicular development in men and no ovulation in women.
After the optimum physical development has been reached HRT for men will continue to ensure that the normal androgen function is maintained; such as libido, muscle development, energy levels, hair growth, and sexual function. In women, a variety of types of HRT will either give a menstruation cycle or not as preferred by the patient. HRT is very important in both men and women to maintain bone density and to reduce the risk of early onset osteoporosis.
The fertility treatments used for both men and women would still include hormone replacement in their action.
There is a range of different preparations available for HRT for both men and women; a lot of these, especially those for women are the same used for standard HRT protocols used when hormone levels fall in later life or after the menopause.
For the men testosterone replacement is achieved either by using daily capsules, daily gel or patches, fortnightly injections, three monthly injections, or six monthly implants. Tablet/capsule forms of HRT rarely give sufficient testosterone levels suitable for men with KS/HH. Different formulations of testosterone are available for treatment which will contain single or multiple forms of testosterone..[33]
The three monthly injection of testosterone undecanoate has become very popular over the past ten years. First produced by the Bayer pharmaceutical company and marketed under the names Nebido, Reandron, or Aveed. In early 2014 Aveed was licensed for use in the US by the Food and Drug Administration (FDA), produced in 3ml vials as opposed the regular 4ml vials used elsewhere around the world.
After the first two injections which are six weeks apart, injections are taken every three months and give good testosterone levels throughout the three-month period with no noticeable tailing-off of levels at the end of the injection cycle. Some patients only require the injection every six months. These injection intervals might be adjusted depending on the response of the individual.
Some treatments may work better with some patients than others so it might be a case of personal choice as which one to use.
There are no specialist HRT treatments available just for women with KS/HH but there are multitude of different HRT products on the market including oral contraceptives and standard post-menopause products. Pills are popular but patches are also available. It may take some trial and error to find the appropriate HRT for the patient depending on how her body reacts to the particular HRT. Specialist medical advice will be required to ensure the correct levels of oestrogen and progesterone are maintained each month, depending on whether the patient requires continuous HRT (no-bleed) or a withdrawal option to create a "menstrual" type bleed. This withdrawal bleed can be monthly or over longer time periods depending on the type of medication used.
Fertility treatments
Fertility treatments for people with KS/HH will require specialist advice from doctors experienced in reproductive endocrinology. There is a good success rate for achieving fertility for patients with KS/HH, with some experts quoting up to a 70% success rate, if IVF techniques are used as well. However, there are factors that can have a negative effect on fertility and specialist advice will be required to determine if these treatments are likely to be successful.
Fertility treatments involve the administration of the gonadotropins LH and FSH in order to stimulate the production and release of eggs and sperm. Women with KS or HH have an advantage over the men as their ovaries normally contain a normal number of eggs and it sometimes only takes a few months of treatment to achieve fertility while it can take males up to two years of treatment to achieve fertility.
A new potential new form of fertility treatment is undergoing clinical trial in 2013 and 2014 by Merck Sharp & Dohme. The trial is evaluating a longer acting form of FSH, in the form of corifollitropin alfa. Injections are taken fortnightly instead of the normal twice weekly it is hoped that this could induce sperm production within months rather than the two years it can take with currently available medications. The trial is expected to last until May 2015.
Human chorionic gonadotrophin (hCG) is sometimes used to stimulate testosterone production in men and ovulation induction in women. For men it acts in the same way as LH; stimulating the Leydig cells in the testes to produce testosterone. Common trade names for hCG products include Pregnyl, Follutein, Profasi, or Choragon. Some men with KS or HH take hCG solely for testosterone production.
Human menopausal gonadotrophin (hMG) is used to stimulate sperm production in men and for multiple egg production and ovulation induction in women. It contains a mixture of both LH and FSH. In men the FSH acts on the sperm producing Sertoli cells in the testes. This can lead to testicular enlargement but can take anything from 6 months to 2 years for an adequate level of sperm production to be achieved. Common trade names for hMG products include Menopur, Menogon, Repronex, or Pergonal.
Purified forms of FSH are also available and are sometimes used with hCG instead of using hMG.
Females with KS / HH would normally require both hCG and FSH in order to achieve fertility. Other cases of female infertility can be treated with just FSH but females (and most males) with KS / CHH would require the use of both forms of gonadotropin injection.
Injections can be intramuscular but are normally taken just underneath the skin (subcutaneous) and are normally taken two or three times a week.
For both men and women, an alternative method (but not widely available), is the use of an infusion pump to provide GnRH (or LHRH) in pulsatile doses throughout the day. This stimulates the pituitary gland to release natural LH and FSH in order to activate testes or ovaries. The use of Kisspeptin delivered in the same pulsatile manner is also under evaluation as a possible treatment for fertility induction.
Epidemiology
The prevalence of idiopathic hypogonadotropic hypogonadism (IHH) and Kallmann syndrome (KS) has been estimated to be in the region of 1 in 10,000 male births.[41] This figure comes from a 1973 study of French Foreign Legion conscripts[15] A more recent paper published in 2011[16] gave the incidence in the Finnish population at 1 in 48,000, with a sex distinction of 1 in 30,000 for males and 1 in 125,000 for females.
Since there is no genetic consensus for the diagnosis of KS and IHH it does make finding a reliable figure for the incidence difficult. It is believed to be between four and five times more common in males than females but there is no obvious genetic reason for this, even though two of the associated gene defects occur on the x-chromosome.
KS and IHH show all versions of genetic inheritance; both x linked or autosomal and dominant or recessive inheritance. Mutations in the KAL-1 gene on the x-chromosome can cause x-linked KS in isolation but other cases of KS and IHH show probable digenic properties, with two gene defects working in combination.
While KS and IHH are normally considered to be congenital conditions other forms have been reported including adult onset IHH and potentially reversible IHH. Cases within the same family do not show the same range symptoms perhaps highlighting the diverse genetic nature of the conditions.
There may also be no obvious family history of inheritance (sporadic or isolated cases), but any case of KS or IHH does have the potential to be passed on to future generations.
Unless there are no accompanying conditions such as heart or neural defects there is normally no effect on life expectancy.
Early onset osteoporosis due to low levels of testosterone or oestrogen can cause problems but otherwise KS and IHH if treated correctly are not associated with a high level of morbidity.
Society and culture
European Consortium
In 2011 a team led by Prof. Nelly Pitteloud and Andrew Dwyer of the University Hospital of Lausanne (CHUV) in Switzerland proposed the formation of a European wide research consortium funded by the European Cooperation on Science and Technology organisation (COST) that would provide a framework for clinicians and researchers to collaborate their research into GnRH deficiency conditions, including Kallmann syndrome and other forms of hypogonadotropic hypogonadism. The first meeting of COST Action BM1105 was held in Brussels in February 2013.
The website (www.gnrhnetwork.eu) of the consortium was launched in March 2013 and will contain information for clinicians, researchers and patients with GnRH deficient conditions. In 2015 the clinical working group published a consensus paper on the diagnosis and treatment of patients with Kallmann syndrome and other forms of GnRH deficiency.[5]
Notable cases
The best-known person who had Kallmann syndrome was probably the jazz vocalist Jimmy Scott; the syndrome was a contributing factor to Scott's unusually high-pitched and distinctive singing voice. In 2004, Canadian writer Brian Brett published a memoir, Uproar's Your Only Music, about growing up with Kallmann syndrome.[42]
Patient perspective
Having Kallmann syndrome can have a profound effect on a person’s life; however, it will affect different people in different ways. Age of diagnosis and treatment is a big key to how well an individual patient copes with the condition. For some patients the ability to put a name to the condition and the knowledge that they are not the only person in the world with this condition is very reassuring.[43] With the key symptom being not going through puberty at the normal age it can produce a huge effect on a person’s social development as well as physical development.
It will vary from person to person but in general men with Kallmann syndrome will have a smaller penile length than the average for the population, which in addition to the lack of testicular development can affect self-confidence to such a degree that sexual activity is not even attempted. Most men with Kallmann syndrome can have a normal, active sex life but the confidence required to achieve this is sometimes beyond some men with Kallmann syndrome and they have less sexual activity than other people the same age.
Another aspect of Kallmann syndrome is social isolation. Since it is such a rare condition, a lot of patients with Kallmann syndrome have never even met or talked to a fellow patient. The ability to meet and talk to other people with the condition goes a long way to helping a patient come to terms with the condition.[44]
Research
Kisspeptin is a protein that regulates the release of GnRH from the hypothalamus, which in turn regulates the release of LH and to a lesser extent, FSH from the anterior pituitary gland. Kisspeptin and its associated receptor ligand GPR54 are known to be involved in the regulation of puberty.
Studies have shown there is potential for Kisspeptin to be used in the diagnosis and treatment of conditions such as Kallmann syndrome and CHH in certain cases.[45][46]
At present there are limitations to the potential therapeutic role of Kisspeptin in cases of Kallmann syndrome and CHH. Studies have shown that GnRH release, and hence LH and FSH response does decrease over time with continuous Kisspeptin administration and there have to be sufficient GnRH releasing neurones present in the hypothalamus in order for the Kisspeptin to be effective. However, this is an ongoing area of research both with Kallmann syndrome and other clinical conditions linked to the dis-regulation of the HPG axis.
A failure of LH and FSH production to Kisspeptin administration over a 24-hour period could be used as a diagnostic marker for Kallmann syndrome and CHH where there is an absence of GnRH releasing neurones.
There does appear to be a role in the use of Kisspeptin in the treatment of acquired amenohrea in women and in some fertility treatments.[47]
The two main international clinical and medical research groups for GnRH deficiency disorders are based at the Reproductive Endocrine Unit at Massachusetts General Hospital in Boston, USA and at the Department of Endocrinology, Diabetes and Metabolism at Centre hospitalier universitaire vaudois, Lausanne, Switzerland headed by Dr William Crowley and Prof. Nelly Pitteloud respectively.
See also
- Isolated hypogonadotropic hypogonadism
- Hypogonadotropic hypogonadism
- Hypogonadism
- Delayed puberty
- Infertility
- Gonadotropin-releasing hormone insensitivity
- Anosmia
References
- 1 2 3 Pitteloud N, Quinton R, Pearce S, Raivio T, Acierno J, et al. (2007). "Digenic mutations account for variable phenotypes in idiopathic hypogonadotropic hypogonadism". J Clin Invest. 117 (2): 457–63. doi:10.1172/JCI29884. PMC 1765517
 . PMID 17235395.
. PMID 17235395. - 1 2 MacColl G, Bouloux P, Quinton R (2002). "Kallmann syndrome: adhesion, afferents, and anosmia". Neuron. 34 (5): 675–8. doi:10.1016/S0896-6273(02)00720-1. PMID 12062015.
- ↑ Schwanzel-Fukuda, M; Bick, D; Pfaff, DW (1989). "Luteinizing hormone-releasing hormone (LHRH)-expressing cells do not migrate normally in an inherited hypogonadal (Kallmann) syndrome.". Brain Res Mol Brain Res. 6 (4): 311–26. doi:10.1016/0169-328x(89)90076-4. PMID 2687610.
- ↑ Valdes-Socin, H (2014). "Reproduction, smell, and neurodevelopmental disorders: genetic defects in different hypogonadotropic hypogonadal syndromes". Frontiers in Endocrinology. 5: 109. doi:10.3389/fendo.2014.00109.
- 1 2 3 Boehm U, Bouloux PM, Dattani MT, et al. (2015). "Expert consensus document: European Consensus Statement on congenital hypogonadotropic hypogonadism-pathogenesis, diagnosis and treatment.". Nat Rev Endocrinol. 11 (Jul 21): 547–64. doi:10.1038/nrendo.2015.112. PMID 26194704.
- 1 2 Guo CY, Jones TH, Eastell R (1997). "Treatment of isolated hypogonadotropic hypogonadism effect on bone mineral density and bone turnover.". J Clin Endocrinol Metab. 82 (2): 658–65. doi:10.1210/jcem.82.2.3758. PMID 9024272.
- ↑ Au MG1, Crowley WF Jr, Buck CL. (2011). "Genetic counseling for isolated GnRH deficiency.". Mol Cell Endocrinol. 346 (1-2): 102–9. doi:10.1016/j.mce.2011.05.041. PMID 21664415.
- 1 2 3 Mitchell AL, Dwyer A, Pitteloud N, Quinton R (2011). "Genetic basis and variable phenotypic expression of Kallmann syndrome: towards a unifying theory.". Trends Endocrinol Metab. 22 (7): 249–58. doi:10.1016/j.tem.2011.03.002. PMID 21511493.
- ↑ Layman L. (2013). "Clinical Testing for Kallmann Syndrome.". J Clin Endocrinol Metab. 98 (5): 1860–1862. doi:10.1210/jc.2013-1624. PMID 23650337.
- ↑ Kallmann FJ, Schönfeld WA, Barrera SE (1943–1944). "The genetic aspects of primary eunuchoidism.". Am J Ment Defic. 48: 203–236.
- ↑ synd/2549 at Who Named It?
- ↑ Maestre de San Juan, Aureliano (1856). "Teratolagia: falta total de los nervios olfactorios con anosmia en un individuo en quien existia una atrofia congenita de los testiculos y miembro viril.". El Siglo Médico. 3: 211–221.
- ↑ Deeb, A.; Robertson, A.; MacColl, G.; Bouloux, P. M. G.; Gibson, M.; Winyard, P. J. D.; Woolf, A. S.; Moghal, N. E.; Cheetham, T. D. (2001). "Multicystic dysplastic kidney and Kallmann's syndrome: a new association?". Nephrology Dialysis Transplantation. 16 (6): 1170–5. doi:10.1093/ndt/16.6.1170. PMID 11390716.
- 1 2 Lee, Peter A.; Houk, Christopher P. (August 13, 2012). "The Smallest Kid in School: Evaluating Delayed Puberty". Medscape Pediatrics.
- 1 2 Fromantin M, et al. (1973). "[Impuberism and hypogonadism at induction into military service. Statistical study].". Probl Actuels Endocrinol Nutr. 16 (May): 179–99. PMID 4147392.
- 1 2 Laitinen EM1, Vaaralahti K, Tommiska J, Eklund E, Tervaniemi M, Valanne L, Raivio T. (2011). "Incidence, phenotypic features and molecular genetics of Kallmann syndrome in Finland.". Orphanet J Rare Dis. 6:41 (Jun 17): 41. doi:10.1186/1750-1172-6-41.
- ↑ Quinton R, Cheow HK, Tymms DJ, Bouloux PM, Wu FC, Jacobs HS (1999). "Kallmann's syndrome: is it always for life?". Clin Endocrinol (Oxf). 50 (4): 481–5. doi:10.1046/j.1365-2265.1999.00708.x. PMID 10468907.
- ↑ Laitinen EM, Tommiska J, Sane T, Vaaralahti K, Toppari J, Raivio T (2012). "Reversible congenital hypogonadotropic hypogonadism in patients with CHD7, FGFR1 or GNRHR mutations.". PLOS ONE. 7 (6): e39450. doi:10.1371/journal.pone.0039450. PMC 3378565. PMID 22724017.
- ↑ Caronia LM, Martin C, Welt CK, Sykiotis GP, et al. (2011). "A genetic basis for functional hypothalamic amenorrhea.". N Engl J Med. 20;364 (3): 215–25. doi:10.1056/NEJMoa0911064. PMC 3045842. PMID 21247312.
- ↑ Soo-Hyun Kim (2015). "Congenital Hypogonadotropic Hypogonadism and Kallmann Syndrome: Past, Present, and Future". Endocrinol Metab (Seoul). 30 (4): 456–466. doi:10.3803/EnM.2015.30.4.456. PMID 4722398.
- ↑ Eckalbar WL, Fisher RE, Rawls A, Kusumi K (2012). "Scoliosis and segmentation defects of the vertebrae.". Wiley Interdiscip Rev Dev Biol. 1 (3): 401–23. doi:10.1002/wdev.34. PMID 23801490.
- 1 2 Bouvattier C, et al. (2011). "Neonatal gonadotropin therapy in male congenital hypogonadotropic hypogonadism.". Nature Reviews Endocrinology. 18;8 (3): 172–82. doi:10.1038/nrendo.2011.164. PMID 22009162.
- ↑ Laitinen EM, Hero M, Vaaralahti K, Tommiska J, Raivio T (2012). "Bone mineral density, body composition and bone turnover in patients with congenital hypogonadotropic hypogonadism.". Int J Androl. 35 (4): 534–40. doi:10.1111/j.1365-2605.2011.01237.x. PMID 22248317.
- ↑ Sidhoum VF, Chan YM, Lippincott MF, et al. (2013). "Reversal and Relapse of Hypogonadotropic Hypogonadism: Resilience and Fragility of the Reproductive Neuroendocrine System.". J. Clin. Endocrinol. Metab. Jan: 861–70. doi:10.1210/jc.2013-2809. PMC 3942233. PMID 24423288.
- ↑ Dwyer AA, Raivio T, Pitteloud N (2016). "MANAGEMENT OF ENDOCRINE DISEASE: Reversible hypogonadotropic hypogonadism.". Eur J Endocrinol. 174 (6): R267–74. doi:10.1530/EJE-15-1033. PMID 26792935.
- 1 2 Oxford Endocrinology Library. Testosterone Deficiency in Men. 2008. ISBN 978-0199545131 Editor: Hugh Jones. Chapter 9. Puberty & Fertility.
- 1 2 Male Hypogonadism. Friedrich Jockenhovel. Uni-Med Science. 2004. ISBN 3-89599-748-X. Chapter 3. Diagnostic work up of hypogonadism.
- ↑ Young J (2012). "Approach to the Male Patient with Congenital Hypogonadotropic Hypogonadism". J Clin Endocrinol Metab. 97 (3): 707–718. doi:10.1210/jc.2011-1664. PMID 22392951.
- ↑ Pitteloud N. (2012). "Managing delayed or altered puberty in boys.". BMJ. 345 (Dec 3): e7913. doi:10.1136/bmj.e7913. PMID 23207503.
- ↑ Bry-Gauillard H, et al. (May 2010). "Congenital hypogonadotropic hypogonadism in females: clinical spectrum, evaluation and genetics.". Ann. Endocrinol. (Paris). 71 (3): 158–162. doi:10.1016/j.ando.2010.02.024. PMID 20363464.
- ↑ Shaw ND, Seminara SB, Welt CK, Au MG, Plummer L, Hughes VA, Dwyer AA, Martin KA, Quinton R, Mericq V, Merino PM, Gusella JF, Crowley WF Jr, Pitteloud N, Hall JE (2011). "Expanding the phenotype and genotype of female GnRH deficiency.". J Clin Endocrinol Metab. 96 (3): E566–76. doi:10.1210/jc.2010-2292. PMID 21209029.
- ↑ Quinton R. (2005). "Adolescent development: advice in ABC of adolescence is potentially misleading.". BMJ. 330 (7494: Apr 2): 789. doi:10.1136/bmj.330.7494.789. PMC 555895. PMID 15802728.
- 1 2 Dunkel L, Quinton R (2014). "Transition in endocrinology: induction of puberty.". Eur J Endocrinol. 170 (6): R229–39. doi:10.1530/EJE-13-0894. PMID 24836550.
- ↑ Yousem DM, Geckle RJ, Bilker W, McKeown DA, Doty RL (1996). "MR evaluation of patients with congenital hyposmia or anosmia". AJR Am J Roentgenol. 166 (2): 439–43. doi:10.2214/ajr.166.2.8553963. PMID 8553963.
- ↑ Dodé C, Teixeira L, Levilliers J, et al. (2006). "Kallmann syndrome: mutations in the genes encoding prokineticin-2 and prokineticin receptor-2". PLoS Genet. 20;2 (10): e175. doi:10.1371/journal.pgen.0020175. PMC 1617130. PMID 17054399.
- ↑ Falardeau J, Chung WC, Beenken A, Raivio T, Plummer L, Sidis Y, Jacobson-Dickman EE, Eliseenkova AV, Ma J, Dwyer A, Quinton R, Na S, Hall JE, Huot C, Alois N, Pearce SH, Cole LW, Hughes V, Mohammadi M, Tsai P, Pitteloud N (2008). "Decreased FGF8 signaling causes deficiency of gonadotropin-releasing hormone in humans and mice.". J Clin Invest. 118 (8): 2822–31. doi:10.1172/JCI34538. PMID 18596921.
- ↑ Kotan, L. D.; Hutchins, B. I.; Ozkan, Y; Demirel, F; Stoner, H; Cheng, P. J.; Esen, I; Gurbuz, F; Bicakci, Y. K.; Mengen, E; Yuksel, B; Wray, S; Topaloglu, A. K. (2014). "Mutations in FEZF1 Cause Kallmann Syndrome". The American Journal of Human Genetics. 95 (3): 326–31. doi:10.1016/j.ajhg.2014.08.006. PMID 25192046.
- 1 2 3 4 5 Miraoui H1, Dwyer AA, Sykiotis GP, Plummer L, Chung W, Feng B, Beenken A, Clarke J, Pers TH, Dworzynski P, Keefe K, Niedziela M, Raivio T, Crowley WF Jr, Seminara SB, Quinton R, Hughes VA, Kumanov P, Young J, Yialamas MA, Hall JE, Van Vliet G, Chanoine JP, Rubenstein J, Mohammadi M, Tsai PS, Sidis Y, Lage K, Pitteloud N. (2013). "Mutations in FGF17, IL17RD, DUSP6, SPRY4, and FLRT3 are identified in individuals with congenital hypogonadotropic hypogonadism.". Am J Hum Genet. 92 (5): 725–43. doi:10.1016/j.ajhg.2013.04.008. PMID 23643382.
- ↑ http://www.endocrine-abstracts.org/ea/0022/ea0022S17.3.htm
- ↑ Han TS, Bouloux PM (2010). "What is the optimal therapy for young males with hypogonadotropic hypogonadism?". Clin Endocrinol (Oxf). 72 (6): 731–7. doi:10.1111/j.1365-2265.2009.03746.x. PMID 19912242.
- ↑ Kallmann Syndrome and Idiopathic Hypogonadotropic Hypogonadism at eMedicine
- ↑ Brian Brett at abcbookworld
- ↑ Chadwick PM, Liao LM, Boyle ME (2005). "Size matters: experiences of atypical genital and sexual development in males.". J Health Psychol. 10 (4): 529–43. doi:10.1177/1359105305053420. PMID 16014390.
- ↑ Smith N, Quinton R (2012). "Kallmann syndrome.". BMJ. 345 e6971 (Dec 3): e6971. doi:10.1136/bmj.e6971. PMID 23207501.
- ↑ Skorupskaite K, George JT, Anderson RA (2014). "The kisspeptin-GnRH pathway in human reproductive health and disease.". Hum Reprod Update. 20 (4): 485–500. doi:10.1093/humupd/dmu009. PMID 24615662.
- ↑ Jyothis T. George; Stephanie B. Seminara. "Kisspeptin and the Hypothalamic Control of Reproduction: Lessons from the Human". Endocrinology. 153 (11): 5130–5136. doi:10.1210/en.2012-1429. PMC 3473216. PMID 23015291.
- ↑ Jayasena CN, Nijher GM, Abbara A, Murphy KG, Lim A, Patel D, Mehta A, Todd C, Donaldson M, Trew GH, et al. (2010). "Twice-weekly administration of kisspeptin-54 for 8 weeks stimulates release of reproductive hormones in women with hypothalamic amenorrhea.". Clin Pharmacol Ther. 88 (6): 840–847. doi:10.1038/clpt.2010.204. PMID 20980998.
External links
- GeneReviews/NCBI/NIH/UW entry on Kallmann syndrome
- Man, 33, seeks puberty", the case of Lawrence Koomson, a physician who was treated for the condition as filmed in the documentary. (BBC)
- Rare Disease UK video on Kallmann syndrome.
- Medline Plus article on hypogonadotropic hypogonadism
- European network for GnRH deficiency conditions. Information for patients, clinicians & researchers