Hydroformylation
| Hydroformylation | |
|---|---|
| Reaction type | Addition reaction |
| Identifiers | |
| RSC ontology ID | RXNO:0000272 |

Hydroformylation, also known as oxo synthesis or oxo process, is an industrial process for the production of aldehydes from alkenes.[1] This chemical reaction entails the net addition of a formyl group (CHO) and a hydrogen atom to a carbon-carbon double bond. This process has undergone continuous growth since its invention in 1938: Production capacity reached 6.6×106 tons in 1995. It is important because aldehydes are easily converted into many secondary products. For example, the resulting aldehydes are hydrogenated to alcohols that are converted to plasticizers or detergents. Hydroformylation is also used in speciality chemicals, relevant to the organic synthesis of fragrances and drugs. The development of hydroformylation, which originated within the German coal-based industry, is considered one of the premier achievements of 20th-century industrial chemistry.
The process typically entails treatment of an alkene with high pressures (between 10 and 100 atmospheres) of carbon monoxide and hydrogen at temperatures between 40 and 200 °C.[2] Transition metal catalysts are required. Invariably, the catalyst dissolves in the reaction medium, i.e. hydroformylation is an example of homogeneous catalysis.
History
The discovery of this reaction is attributed to Otto Roelen, who was investigating the Fischer-Tropsch reaction (F-T). Aldehydes and diethylketone were obtained when ethylene was added to an F-T reactor. Through these studies, Roelen discovered the involvement of cobalt catalysts. HCo(CO)4, which had been isolated prior to Roelen, was shown to be an excellent catalyst.[3][4] The term oxo synthesis was created by the Ruhrchemie patent department, who expected the process to be applicable to the preparation of both aldehydes and ketones. Subsequent work demonstrated that the ligand tributylphosphine (PBu3) improved the selectivity of the cobalt-catalysed process.
In the 1960s, highly active rhodium catalysts were discovered.[5] Since the 1970s, most hydroformylation relies on catalysts based on rhodium.[6] Subsequent research led to the development of water-soluble catalysts that facilitate the separation of the products from the catalyst.[7]
Mechanism
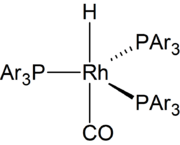
The overall mechanism resembles that for homogeneous hydrogenation with additional steps. The reaction begins with the generation of coordinatively unsaturated metal hydrido carbonyl complex such as HCo(CO)3 and HRh(CO)(PPh3)3. Such species bind alkenes, and the resulting complex undergoes a migratory insertion reaction to form an alkyl complex.
Selectivity
A key consideration of hydroformylation is the "normal" vs. "iso" selectivity. For example, the hydroformylation of propylene can afford two isomeric products, butyraldehyde or isobutyraldehyde:
- H2 + CO + CH3CH=CH2 → CH3CH2CH2CHO ("normal")
- vs.
- H2 + CO + CH3CH=CH2 → (CH3)2CHCHO ("iso")
These isomers result from the differing ways of inserting the alkene into the M–H bond. Of course, both products are not equally desirable. Much research was dedicated to the quest for catalyst that favored the normal isomer.
Steric effects
When the hydrogen is transferred to the carbon bearing the most hydrogen atoms (Markovnikov addition) the resulting alkyl group has a larger steric bulk close to the ligands on the cobalt. If the ligands on the cobalt are bulky (such as tributyl phosphine), then this steric effect is greater. Hence, the mixed carbonyl/phosphine complexes offer a greater selectivity toward the straight chain products.

Electronic effects
In addition, the more electron-rich the hydride complex is the less proton-like the hydride is. Thus, as a result, the electronic effects that favour the Markovnikov addition to an alkene are less able to direct the hydride to the carbon atom bearing the most hydrogens already. Thus, as a result, as the metal centre becomes more electron-rich, the catalyst becomes more selective for the straight chain compounds.
Acetyl formation
After the alkyl formation a second migatory insertion converts the alkyl into an acetyl ligand (this is when the alkyl carbon forms a bond with the carbon of a carbonyl ligand). The vacant site on the metal is filled by two hydrogens (from the oxidative insertion of a hydrogen molecule. One of these hydrides then takes part in a reductive elimination to form the molecule of the aldehyde and the complex [HCo(CO)3].

It is important that the rate of migatory insertion of the carbonyl into the carbon-metal bond of the alkyl is fast; in systems where the migatory insertion does not occur (such as nickel hydride tristriphenyl phosphite), the reaction of the hydride with the alkene is reversible. This results in the isomerisation of the alkene, in this way oct-2-ene could be converted into a mixture of both oct-1-ene and oct-2-ene by a beta hydride elimination from the alkyl. In the system below, the rate of insertion of the carbonyl carbon into the C-M bond is likely to be greater than the rate of beta-hydride elimination. If the converse was true then some n-C8H17CHO would have been formed.[8] Hydroformylation of 2-octene: the rhodium catalyst is coordinated to acac and carbon monoxide and encapsulated in a molecular self-assembly process by zinc tetraphenylporphyrin or Zn-tpp and the pyridine analogue of triphenylphosphine. In this process very much like the way enzymes work encapsulation of the catalytic site explains the observed regioselectivity:

Asymmetric hydroformylation
Hydroformylation of prochiral alkenes creates new stereocenters. Using chiral phosphine ligands, the hydroformylation can be tailored to favor one enantiomer.[9] Thus, for example, Dexibuprofen, the (+)−(s)-enantiomer of Ibuprofen, can be produced by enantioselective hydroformylation followed by oxidation.
Industrial processes
The industrial processes vary depending on the chain length of the olefin to be hydroformylated, the catalyst metal and ligands, and the recovery of the catalyst. The original Ruhrchemie process produced propanal from ethene and syngas using cobalt tetracarbonyl hydride. Today, industrial processes based on cobalt catalysts are mainly used for the production of medium- to long-chain olefins, whereas the rhodium-based catalysts are usually used for the hydroformylation of propene. The rhodium catalysts are significantly more expensive than cobalt catalysts. In the hydroformylation of higher molecular weight olefins the separation of the catalyst from the produced aldehydes is difficult.
BASF-oxo process
The BASF-oxo process starts mostly with higher olefins and relies on cobalt carbonyl-based catalyst.[10] By conducting the reaction at low temperatures, one observes increased selectivity favoring the linear product. The process is carried out at a pressure of about 30 MPa and in a temperature range of 150 to 170 °C. The cobalt is recovered from the liquid product by oxidation to water-soluble Co2 +, followed by the addition of aqueous formic or acetic acids. This process gives an aqueous phase of cobalt, which can then recycled. Losses are compensated by the addition of cobalt salts.[11]
Exxon process
The Exxon process, also Kuhlmann- or PCUK – oxo process, is used for the hydroformylation of C6–C12 olefins. The process relies on cobalt catalysts. In order to recover the catalyst, an aqueous sodium hydroxide solution or sodium carbonate is added to the organic phase. By extraction with olefin and neutralization by addition of sulfuric acid solution under carbon monoxide pressure the metal carbonyl hydride can recovered. This is stripped out with syngas, absorbed by the olefin, and returned to the reactor. Similar to the BASF process, the Exxon process is carried out at a pressure of about 30 MPa and at a temperature of about 160 to 180 °C.[11]
Shell process
The Shell process uses cobalt complexes modified with phosphine ligands for the hydroformylation of C7–C14 olefins. The resulting aldehydes are directly hydrogenated to the fatty alcohols, which are separated by distillation, which allows the catalyst to be recycled. The process has good selectivity to linear products, which find use as feedstock for detergents. The process is carried out at a pressure of about 4 to 8 MPa and at a temperature range of about 150–190 °C.[11]
Union Carbide process
The Union Carbide (UCC) process, also known as low-pressure oxo process (LPO), relies on a rhodium catalyst dissolved in high-boiling thick oil, a higher molecular weight condensation product of the primary aldehydes, for the hydroformylation of propene. The reaction mixture is separated in a falling film evaporator from volatile components. The liquid phase is distilled and butyraldehyde is removed as head product while the catalyst containing bottom product is recycled to the process. The process is carried out at about 1.8 MPa and 95–100 °C.[11]
Ruhrchemie/Rhone–Poulenc process

The Ruhrchemie/Rhone–Poulenc process (RCRPP) relies on a rhodium catalyst with water-soluble TPPTS as ligand (Kuntz Cornils catalyst) for the hydroformylation of propene.[12] The tri-sulfonation of triphenylphosphane ligand provides hydrophilic properties to the organometallic complex. The catalyst complex carries nine sulfonate-groups and is highly soluble in water (about 1 kg L−1), but not in the emerging product phase.[13] The water-soluble TPPTS is used in about 50-fold excess, whereby the leaching of the catalyst is effectively suppressed. Reactants are propene and syngas consisting of hydrogen and carbon monoxide in a ratio of 1.1:1. A mixture of butyraldehyde and isobutyraldehyde in the ratio 96:4 is generated with few by-products such as alcohols, esters and higher boiling fractions.[13] The Ruhrchemie/Rhone-Poulenc-process is the first commercially available two-phase system in which the catalyst is present in the aqueous phase. In the progress of the reaction an organic product phase is formed which is separated continuously by means of phase separation, wherein the aqueous catalyst phase remains in the reactor.[13]
The process is carried out in a stirred tank reactor where the olefin and the syngas are bubbled from the bottom of the reactor through the catalyst phase under intensive stirring. The resulting crude aldehyde phase is separated at the top from the aqueous phase. The aqueous catalyst-containing solution is re-heated via a heat exchanger and pumped back into the reactor.[13] The excess olefin and syngas is separated from the aldehyde phase in a stripper and fed back to the reactor. The generated heat is used for the generation of process steam, which is used for subsequent distillation of the organic phase to separate into butyraldehyde and isobutyraldehyde.[13] Potential catalyst poisons coming from the synthesis gas migrate into the organic phase and removed from the reaction with the aldehyde. Thus there is no accumulation of catalyst poisons, and the elaborate fine purification of the syngas can be omitted.[13]
A plant was built in Oberhausen in 1984, which was debottlenecked in 1988 and again in 1998 up to a production capacity of 500,000 t/a butanal. The conversion rate of propene is 98% and the selectivity to the n-butanal is high. During the life time of a catalyst batch in the process less than 1 ppb rhodium is lost.[14]
Other substrates
Cobalt carbonyl and rhodium complexes catalyse the hydroformylation of formaldehyde and ethylene oxide to give 2-hydroxyacetaldehyde and 3-hydroxypropanaldehyde, which can then be hydrogenated to ethylene glycol and 1,3-propanediol, respectively. The reactions work best when the solvent is basic (such as pyridine).[15][16]

In the case of dicobalt octacarbonyl or Co2(CO)8 as a catalyst, 3-pentanone can arise from ethylene and CO, in the absence of hydrogen. A proposed intermediate is the ethylene-propionyl species [CH3C(O)Co(CO)3(ethylene)] which undergoes a migratory insertion to form [CH3COCH2CH2Co(CO)3]. The required hydrogen arises from the water shift reaction. For details, see[17]

If the water shift reaction is not operative, the reaction affords a polymer containing alternating carbon monoxide and ethylene units. Such aliphatic polyketones are more conventionally prepared using palladium catalysts.[18]
In addition to pure olefins, functionalized olefins such as allyl alcohol can be hydroformylated. The target product 1,4-butanediol and its isomer is obtained with isomerization free catalysts such as rhodium-triphenylphosphine complexes. The use of the cobalt complex leads by isomerization of the double bond to n-propanal.[19] The hydroformylation of alkenyl ethers and alkenyl esters occurs usually in the α-position to the ether or ester function.
The hydroformylation of acrylic acid and methacrylic acid leads in the rhodium-catalyzed process in the first step to the Markovnikov product.[20] By variation of the reaction conditions the reaction can be directed to different products. A high reaction temperature and low carbon monoxide pressure favors the isomerization of the Markovnikov product to the thermodynamically more stable β-isomer, which leads to the n-aldehyde. Low temperatures and high carbon monoxide pressure and an excess of phosphine, which blocks free coordination sites, can lead to faster hydroformylation in the α-position to the ester group and suppress the isomerization.[20]
Side- and consecutive reactions
Olefins
Side reactions of the olefins are the isomerization and hydrogenation of the olefinic double bond. While the alkanes resulting from hydrogenation of the double bond do not participate further in the reaction, the isomerization of the double bond with subsequent formation of the n-alkyl complexes is a desired reaction. The hydrogenation is usually of minor importance; However, Cobalt-phosphine-modified catalysts can have an increased hydrogenation activity, where up to 15% of the olefin is hydrogenated.
Aldehydes
A usually desired consecutive reaction is the hydrogenation of the aldehydes to alcohols. Higher temperatures and hydrogen partial pressures favor the hydrogenation of the resulting aldehyde to the alcohol. For the reaction mechanism it is believed that the aldehyde initially forms a CO-π-complex with the catalyst. This is rearranged to the alkoxide complex and by subsequent oxidative addition of hydrogen the alcohol is eliminated and the starting complex is formed.
The aldehydic carbon-oxygen double bond can also be subject to hydroformylation, which leads to formic acid and its esters. The reaction requires the carbon monoxide insertion into the oxygen-metal bond of the alkoxide complex. The resulting Formyl complex can converted into the formic acid esters and the starting complex by oxidative addition of hydrogen. The initially produced aldehydes can react further by aldol condensation to either target product precursors like 2-ethylhexene-al or higher molecular weight condensation products, so-called thick oil.
Catalyst and ligands
The triphenylphosphine complexes are under reaction conditions potentially subject to hydrogenation and can release benzene. The insertion of carbon monoxide in an intermediate metal-phenyl bond can lead to the formation of benzaldehyde or by subsequent hydrogenation to benzyl alcohol.[21] One of the ligands phenyl-groups can be replaced by propene, and the resulting diphenylpropylphosphine ligand can inhibit the hydroformylation reaction due to its increased basicity.[21]
Trace impurities of the starting materials with oxygen or sulfur and their compounds can contribute to the oxidation of phosphorus (III) – to phosphorus (V) compounds or lead to catalytically inactive metal oxides and – sulfides.
References
- ↑ Ojima, I.; Tsai, C.-Y.; Tzamarioudaki, M.; Bonafoux, D. Org. React. 2000, 56, 1. doi:10.1002/0471264180.or056.01
- ↑ Pino, P.; Botteghi, C. (1977). "Aldehydes from olefins: cyclohexanecarboxaldehyde". Organic Syntheses. 57: 11. doi:10.15227/orgsyn.057.0011.
- ↑ Boy Cornils; Wolfgang A. Herrmann; Manfred Rasch (1994). "Otto Roelen, Pioneer in Industrial Homogeneous Catalysis". Angewandte Chemie International Edition in English. 33 (21): 2144–2163. doi:10.1002/anie.199421441.
- ↑ http://www.tu-braunschweig.de/Medien-DB/anchem/homogenekat-4bw.pdf
- ↑ Evans, D., Osborn, J. A., Wilkinson, G. (1968). "Hydroformylation of Alkenes by Use of Rhodium Complex Catalyst" (PDF). Journal of the Chemical Society. 33 (21): 3133–3142. doi:10.1039/J19680003133.
- ↑ J. F. Hartwig; Organotransition metal chemistry – from bonding to catalysis. University Science Books. 2009. 753, 757–578. ISBN 978-1-891389-53-5.
- ↑ Cornils, B.; Herrmann, W. A. (eds.) “Aqueous-Phase Organometallic Catalysis” VCH, Weinheim: 1998
- ↑ High-Precision Catalysts: Regioselective Hydroformylation of Internal Alkenes by Encapsulated Rhodium Complexes Kuil, M.; Soltner, T.; van Leeuwen, P. W. N. M.; Reek, J. N. H. Journal of the American Chemical Society; 2006; 128(35) pp 11344–45; doi:10.1021/ja063294i
- ↑ Watkins, A. L.; Hashiguchi, B. G.; Landis, C. R., Highly Enantioselective Hydroformylation of Aryl Alkenes with Diazaphospholane Ligands. Organic Letters 2008, 10 (20), 4553–4556.
- ↑ G. Duembgen, D. Neubauer: Grosstechnische Herstellung von Oxo-Alkoholen aus Propylen in der BASF. In: Chemie Ingenieur Technik – CIT. 41, 1969, S. 974–80, doi:10.1002/cite.330411708.
- 1 2 3 4 Boy Cornils, Wolfgang A. Herrmann, Chi-Huey Wong, Horst -Werner Zanthoff: Catalysis from A to Z: A Concise Encyclopedia, 2408 Seiten, Verlag Wiley-VCH Verlag GmbH & Co. KGaA, (2012), ISBN 3-527-33307-X.
- ↑ W. A. Herrmann, C. W. Kohlpaintner, Angew. Chem. 1993, 105, 1588.
- 1 2 3 4 5 6 Ernst Wiebus, Boy Cornils: Die großtechnische Oxosynthese mit immobilisiertem Katalysator. In: Chemie Ingenieur Technik. 66, 1994, S. 916–923, doi:10.1002/cite.330660704.
- ↑ Manfred Baerns, Arno Behr, Axel Brehm, Jürgen Gmehling, Hanns Hofmann, Ulfert Onken: Technische Chemie Lehrbuch. 480 Abbildungen, 190 Tabellen. Wiley VCH Verlag GmbH, September 2006, ISBN 3-527-31000-2.
- ↑ Chan A.S.C.; Shieh H-S. (1994). "A mechanistic study of the homogeneous catalytic hydroformylation of formaldehyde: synthesis and characterization of model intermediates". Inorganica Chimica Acta. 218 (1–2): 89–95. doi:10.1016/0020-1693(94)03800-7.
- ↑ Spencer A. (1980). "Hydroformylation of formaldehyde catalysed by rhodium complexes". Journal of Organometallic Chemistry. 194 (1–2): 113–123. doi:10.1016/S0022-328X(00)90343-7.
- ↑ Murata K.; Matsuda A. (1981). "Application of Homogeneous Water-Gas Shift Reaction III Further Study of the Hydrocarbonylation – A highly Selective Formation of Diethyl Keton from Ethene, CO and H2O". Bulletin of the Chemical Society of Japan. 54 (7): 2089–2092. doi:10.1246/bcsj.54.2089.
- ↑ J. Liu; B.T. Heaton; J.A. Iggo; R. Whyman (2004). "The Complete Delineation of the Initiation, Propagation, and Termination Steps of the Carbomethoxy Cycle for the Carboalkoxylation of Ethene by Pd–Diphosphane Catalysts". Angew. Chem. Int. Ed. 43: 90–94. doi:10.1002/anie.200352369.
- ↑ Bernhard Fell, Wolfgang Rupilius, Friedrich Asinger: Zur Frage der Isomerenbildung bei der Hydroformylierung höhermolekularer Olefine mit komplexen Kobalt- und Rhodiumkatalysatoren. In: Tetrahedron Letters. 9, 1968, S. 3261–3266, doi:10.1016/S0040-4039(00)89542-8.
- 1 2 Jürgen Falbe, Ch. R. Adams: Carbon Monoxide in Organic Synthesis, Springer Verlag, 1970, ISBN 3-540-04814-6.
- 1 2 Arno Behr: Angewandte homogene Katalyse, Wiley-VCH. Weinheim, ISBN 3-527-31666-3.
Further reading
- “Applied Homogeneous Catalysis with Organometallic Compounds: A Comprehensive Handbook in Two Volumes (Paperback) by Boy Cornils (Editor), W. A. Herrmann (Editor). ISBN 3-527-29594-1
- “Rhodium Catalyzed Hydroformylation” P. W. N. M. van Leeuwen, C. Claver Eds.; Springer; (2002). ISBN 1-4020-0421-4
- “Homogeneous Catalysis: Understanding the Art” by Piet W. N. M. van Leeuwen Springer; 2005. ISBN 1-4020-3176-9
- Imyanitov N.S./ Hydroformylation of Olefins with Rhodium Complexes // Rhodium Express. 1995. No 10–11 (May). pp. 3–62 (Eng). ISSN 0869-7876