Hurler syndrome
| Hurler syndrome | |
|---|---|
| Classification and external resources | |
| ICD-10 | E76.0 |
| ICD-9-CM | 277.5 |
| OMIM | 607014 |
| DiseasesDB | 6067 |
| MedlinePlus | 001204 |
| eMedicine | ped/1031 |
| MeSH | D008059 |
| GeneReviews | |
Hurler syndrome, also known as mucopolysaccharidosis type I (MPS I), Hurler's disease, also gargoylism, is a genetic disorder that results in the buildup of glycosaminoglycans (formerly known as mucopolysaccharides) due to a deficiency of alpha-L iduronidase, an enzyme responsible for the degradation of mucopolysaccharides in lysosomes.[1]:544 Without this enzyme, a buildup of heparan sulfate and dermatan sulfate occurs in the body. Symptoms appear during childhood and early death can occur due to organ damage.
MPS I is divided into three subtypes based on severity of symptoms. All three types result from an absence of, or insufficient levels of, the enzyme α-L-iduronidase. MPS I H or Hurler syndrome is the most severe of the MPS I subtypes. The other two types are MPS I S or Scheie syndrome and MPS I H-S or Hurler-Scheie syndrome.
Hurler syndrome is often classified as a lysosomal storage disease, and is clinically related to Hunter Syndrome.[2] Hunter syndrome is X-linked while Hurler syndrome is autosomal recessive.
It is named for Gertrud Hurler (1889–1965), a German pediatrician.[3][4]
Prevalence
Hurler syndrome has an overall frequency of 1 per 100,000.[5] The mucopolysaccharidoses as a whole have a frequency of 1 in every 25,000 births.
Genetics
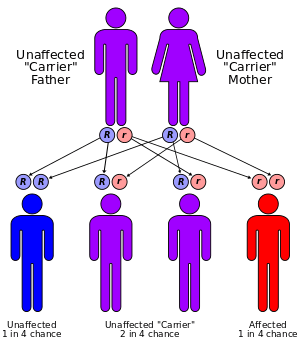
Children born to an MPS I parent carry a defective IDUA gene, which has been mapped to the 4p16.3 site on chromosome 4. The gene is named IDUA because of its iduronidase enzyme protein product. As of 2001, 52 different mutations in the IDUA gene have been shown to cause Hurler syndrome.
Because Hurler syndrome is an autosomal recessive disorder, affected persons have two non-working copies of the IDUA gene. If someone is born with one normal and one defective copy of the gene (s)he is called a carrier and will produce less α-L-iduronidase than an individual with two normal copies of the gene. The slightly reduced production of the enzyme in carriers, however, remains sufficient for normal function and the person should not show any symptoms of the disease.
Features
The condition is marked by progressive deterioration, hepatosplenomegaly, dwarfism and unique facial features. There is a progressive mental retardation, with death frequently occurring by the age of 10 years.[6]
Developmental delay is evident by the end of the first year, and patients usually stop developing between ages 2 and 4. This is followed by progressive mental decline and loss of physical skills. Language may be limited due to hearing loss and an enlarged tongue. In time, the clear layers of the cornea become clouded and retinas may begin to degenerate. Carpal tunnel syndrome (or similar compression of nerves elsewhere in the body) and restricted joint movement are common.
Affected children may be large at birth and appear normal but may have inguinal (in the groin) or umbilical (where the umbilical cord passes through the abdomen) hernias. Growth in height may be initially faster than normal, then begins to slow before the end of the first year and often ends around age 3. Many children develop a short body trunk and a maximum stature of less than 4 feet. Distinct facial features (including flat face, depressed nasal bridge, and bulging forehead) become more evident in the second year. By age 2, the ribs have widened and are oar-shaped. The liver, spleen and heart are often enlarged. Children may experience noisy breathing and recurring upper respiratory tract and ear infections. Feeding may be difficult for some children, and many experience periodic bowel problems. Children with Hurler syndrome often die before age 10 from obstructive airway disease, respiratory infections, or cardiac complications.
Diagnosis
Diagnosis often can be made through clinical examination and urine tests (excess mucopolysaccharides are excreted in the urine). Enzyme assays (testing a variety of cells or body fluids in culture for enzyme deficiency) are also used to provide definitive diagnosis of one of the mucopolysaccharidoses. Prenatal diagnosis using amniocentesis and chorionic villus sampling can verify if a fetus either carries a copy of the defective gene or is affected with the disorder. Genetic counseling can help parents who have a family history of the mucopolysaccharidoses determine if they are carrying the mutated gene that causes the disorders.
Treatment
Enzyme replacement therapies are currently in use. BioMarin Pharmaceutical provides therapeutics for mucopolysaccaradosis type I (MPS I), by manufacturing laronidase (Aldurazyme), commercialized by Genzyme. Enzyme replacement therapy has proven useful in reducing non-neurological symptoms and pain.
Bone marrow transplantation (BMT) and umbilical cord blood transplantation (UCBT) can be used as treatments for MPS. Abnormal physical characteristics, except for those affecting the skeleton and eyes, can be improved, and neurologic degeneration can often be halted. BMT and UCBT are high-risk procedures with high rates of morbidity and mortality. There is no cure for MPS I.
Gene therapy
There is currently a great deal of interest in treating MPS I with gene therapy. This approach has been taken with retroviral, lentiviral, AAV, and even nonviral vectors to deliver the iduronidase gene. Successful treatment of the mouse, dog, and cat models of MPS I has occurred and may pave the way for future human trials.
Prognosis
A large British study from 2008 found a median estimated life expectancy of 11.6 years.[6]
See also
References
- ↑ James, William D.; Berger, Timothy G.; et al. (2006). Andrews' Diseases of the Skin: clinical Dermatology. Saunders Elsevier. ISBN 0-7216-2921-0.
- ↑ http://www.ninds.nih.gov/disorders/mucopolysaccharidoses/detail_mucopolysaccharidoses.htm
- ↑ Hurler's syndrome at Who Named It?
- ↑ Hurler, G. (1919). "Über einen Typ multipler Abartungen, vorwiegend am Skelettsystem". Zeitschrift für Kinderheilkunde. Berlin. 24: 220–234.
- ↑ Mucopolysaccharidosis Type I at eMedicine
- 1 2 Moore, David; Connock, Martin J.; Wraith, Ed; Lavery, Christine (2008-01-01). "The prevalence of and survival in Mucopolysaccharidosis I: Hurler, Hurler-Scheie and Scheie syndromes in the UK". Orphanet Journal of Rare Diseases. 3: 24. doi:10.1186/1750-1172-3-24. ISSN 1750-1172. PMC 2553763
 . PMID 18796143.
. PMID 18796143.
External links
- National MPS Society
- GeneReview/NIH/UW entry on MucopolysDaccharidosis Type I
- Hurler Syndrome Community
- Genetic Targeting of the Albumin Locus to Treat Hemophilia