Holt–Oram syndrome
| Holt-Oram syndrome | |
|---|---|
| Classification and external resources | |
| Specialty | medical genetics |
| ICD-10 | Q87.2 |
| OMIM | 142900 |
| DiseasesDB | 5988 |
| eMedicine | med/2940 ped/1021 |
| GeneReviews | |
Holt–Oram syndrome (also called Heart and Hand Syndrome, atrio-digital syndrome, atriodigital dysplasia, cardiac-limb syndrome, heart-hand syndrome type 1, HOS, ventriculo-radial syndrome[1]) is an autosomal dominant disorder that affects bones in the arms and hands (the upper limbs) and may also cause heart problems. The syndrome includes an absent radial bone in the arms, an atrial septal defect, and a first degree heart block.[2] Holt–Oram syndrome is considered to be a phenocopy of thalidomide, since both the genetic disorder and the teratogenic effects have similar presentations in individuals.
Presentation
All people with this disorder have at least one limb abnormality that affects bones in the wrist (carpal bones). Often, these wrist bone abnormalities can be detected only by X-ray. Affected individuals may have additional bone abnormalities that can include polydactyly, a hypoplastic thumb or a Triphalangeal thumb, partial or complete absence of bones in the forearm, an underdeveloped Humerus, and abnormalities that affect the Clavicle and Scapula. Bone abnormalities may affect each arm differently, and the left side can be affected more than the right side. In some cases, only one arm and/or hand is affected.
About 75 percent of individuals with Holt–Oram syndrome have heart problems. The most common problem is a defect in the muscular wall, or septum, that separates the right and left sides of the heart (atria).[3] Atrial septal defects (ASD) are caused by a hole in the septum between the left and right upper chambers of the heart (atria), and ventricular septal defects (VSD) are caused by a hole in the septum between the left and right lower chambers of the heart (ventricles). Sometimes people with Holt–Oram syndrome have cardiac conduction disease, which is caused by abnormalities in the electrical system that coordinates contractions of the heart chambers. Cardiac conduction disease can lead to problems such as a slow heart rate (bradycardia) or a rapid and ineffective contraction of the heart muscles (fibrillation). Cardiac conduction disease can occur along with other heart defects (such as septal defects) or as the only heart problem in people with Holt–Oram syndrome.
Genetics
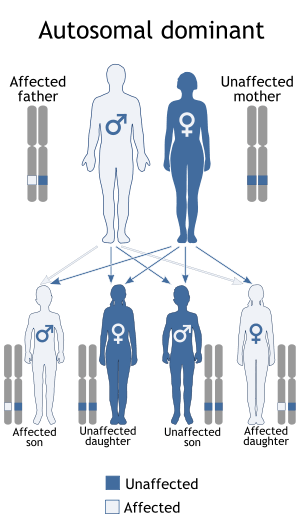
Mutations in the TBX5 gene cause Holt–Oram syndrome.[4][5] The TBX5 gene plays a role in the development of the heart and upper limbs before birth. In particular, this gene appears to be important for the process that divides the developing heart into four chambers (cardiac septation). The TBX5 gene also appears to play a critical role in regulating the development of bones in the arm and hand. Mutations in this gene probably disrupt the development of the heart and upper limbs, leading to the characteristic features of Holt–Oram syndrome.
Holt–Oram syndrome is considered an autosomal dominant disorder. This means the defective gene is located on an autosome, and only one copy of the gene, inherited from a parent who has the disorder, is sufficient to cause the disorder.
Other cases of Holt–Oram syndrome are sporadic, and result from new mutations in the TBX5 gene that occur in people with no history of the disorder in their family. Holt–Oram syndrome is estimated to affect 1 in 100,000 individuals.
In some cases, Holt-Oram has a multiplier effect when passed on generation to generation. An affected child of an affected parent will likely face greater challenges than the parent did. In rare cases, some carriers are unable to reproduce at all due to the severity of the condition.
History
It is named for Mary Holt and Samuel Oram who discovered first published a paper on it in 1960.[6][7]
See also
References
- ↑ "Holt-Oram syndrome". Genetics Home Reference. National Institute of Health, USA.
- ↑ Skelley, Tao Le, Vikas Bhushan, Nathan William. First aid for the USMLE step 2 CK (8th ed.). New York: McGraw-Hill Medical. p. 357. ISBN 978-0-07-176137-6.
- ↑ Bossert, T; Walther, T; Gummert, J; Hubald, R; Kostelka, M; Mohr, FW (October 2002). "Cardiac malformations associated with the Holt-Oram syndrome—report on a family and review of the literature.". The Thoracic and cardiovascular surgeon. 50 (5): 312–4. doi:10.1055/s-2002-34573. PMID 12375192. Retrieved 7 November 2012.
- ↑ Li QY, Newbury-Ecob RA, Terrett JA, et al. (January 1997). "Holt-Oram syndrome is caused by mutations in TBX5, a member of the Brachyury (T) gene family". Nat. Genet. 15 (1): 21–9. doi:10.1038/ng0197-21. PMID 8988164.
- ↑ Basson CT, Bachinsky DR, Lin RC, et al. (January 1997). "Mutations in human TBX5 [corrected] cause limb and cardiac malformation in Holt-Oram syndrome". Nat. Genet. 15 (1): 30–5. doi:10.1038/ng0197-30. PMID 8988165.
- ↑ Virdis, G.; Dessole, M.; Dessole, S.; Ambrosini, G.; Cosmi, E.; Cherchil, P. L.; Capobianco, G. (2016-01-01). "Holt Oram syndrome: a case report and review of the literature". Clinical and Experimental Obstetrics & Gynecology. 43 (1): 137–139. ISSN 0390-6663. PMID 27048037.
- ↑ Holt, Mary; Oram, Samuel (1960-04-01). "FAMILIAL HEART DISEASE WITH SKELETAL MALFORMATIONS". British Heart Journal. 22 (2): 236–242. ISSN 0007-0769. PMC 1017650
 . PMID 14402857.
. PMID 14402857.
External links
- GeneReview/NIH/UW entry on Holt-Oram Syndrome
- Holt–Oram syndrome at NLM Genetics Home Reference
- Holt-Oram syndrome: Encyclopedia of Genetic Disorders
- http://holt-oram.ning.com/ social network site for those affected by the condition