Harderoporphyria
| Harderoporphyria | |
|---|---|
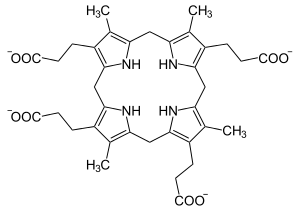 | |
| Coproporphyrinogen III | |
| Classification and external resources | |
| OMIM | 121300 |
Harderoporphyria is a rare disorder of heme biosynthesis, inherited in an autosomal recessive manner caused by specific mutations in the CPOX gene. Mutations in CPOX usually cause hereditary coproporphyria, an acute hepatic porphyria, however the K404E mutation in a homozygous or compound heterozygous state with a null allele cause the more severe harderoporphyria.[1] Harderoporphyria is the first known metabolic disorder where the disease phenotype depended on the type and location of the mutations in a gene associated with multiple disorders.[2]
In contrast with other porphyrias, which typically present with either cutaneous lesions after exposure to sunlight or acute neurovisceral attack at any age (most commonly in adulthood), harderoporphyria is characterized by jaundice, anemia enlarged liver and spleen, often presenting in the neonatal period. Later in life, these individuals may present with photosensitivity similar to that found in cutaneous porphyrias.[2]
Biochemically, harderoporphyria presents with a distinct pattern of increased harderoporphyrin (2-vinyl-4,6,7-tripropionic acid porphyrin)[3] in urine and particularly in feces, a metabolite that is not seen in significant quantities in any other porphyria.[2] Enzyme tests show markedly reduced activity of coproporphyrinogen oxidase, compared to both unaffected individuals and those affected with hereditary coproporphyria, consistent with recessive inheritance.[2]
Harderoporphyria is a rare condition, with less than 10 cases reported worldwide. It may be underdiagnosed, as it does not have the typical presentation associated with a porphyria.[2] It was identified as a variant type of coproporphyria in 1983, in a family with three children identified at birth with jaundice and hemolytic anemia.[4] There is no standard treatment for harderoporphyria; care is mainly focused on the management of symptoms.[4][5]
References
- ↑ Schmitt, C.; Gouya, L.; Malonova, E.; Lamoril, J.; Camadro, J. M.; Flamme, M.; Rose, C.; Lyoumi, S.; Da Silva, V.; Boileau, C.; Grandchamp, B.; Beaumont, C.; Deybach, J. C.; Puy, H. (2005). "Mutations in human CPO gene predict clinical expression of either hepatic hereditary coproporphyria or erythropoietic harderoporphyria". Human Molecular Genetics. 14 (20): 3089–3098. doi:10.1093/hmg/ddi342. PMID 16159891.
- 1 2 3 4 5 "#121300 COPROPORPHYRIA, HEREDITARY; HCP". Johns Hopkins University. Retrieved 2012-05-27.
- ↑ Gorchein, A.; Danton, M.; Lim, C. K. (October 2005). "Harderoporphyrin: a misnomer". Biomedical Chromatography. pp. 565–569. doi:10.1002/bmc.480.
- 1 2 Nordmann, Y.; Grandchamp, B.; De Verneuil, H.; Phung, L.; Cartigny, B.; Fontaine, G. (1983). "Harderoporphyria: A variant hereditary coproporphyria". Journal of Clinical Investigation. 72 (3): 1139–1149. doi:10.1172/JCI111039. PMC 1129282
 . PMID 6886003.
. PMID 6886003. - ↑ Lamoril, J.; Puy, H.; Gouya, L.; Rosipal, R.; Da Silva, V.; Grandchamp, B.; Foint, T.; Bader-Meunier, B.; Dommergues, J. P.; Deybach, J. C.; Nordmann, Y. (1998). "Neonatal hemolytic anemia due to inherited harderoporphyria: Clinical characteristics and molecular basis". Blood. 91 (4): 1453–1457. PMID 9454777.