GTPase-activating protein
GTPase-Activating Proteins, or GAPs, or GTPase-Accelerating Proteins are a family of regulatory proteins whose members can bind to activated G proteins and stimulate their GTPase activity, with the result of terminating the signaling event.[1] GAPs are also known as RGS protein, or RGS proteins,[2] and these proteins are crucial in controlling the activity of G proteins. Regulation of G proteins is important because these proteins are involved in a variety of important cellular processes. The large G proteins, for example, are involved in transduction of signaling from the G protein-coupled receptor for a variety of signaling processes like hormonal signaling,[2] and small G proteins are involved in processes like cellular trafficking and cell cycling.[3] GAP’s role in this function is to turn the G protein’s activity off. In this sense, GAPs function is opposite to that of guanine nucleotide exchange factors (GEFs), which serve to enhance G protein signaling.[4]
Mechanism
GAP are heavily linked to the G-protein linked receptor family. The activity of G proteins comes from their ability to bind guanosine triphosphate (GTP). Binding of GTP inherently changes the activity of the G proteins and increases their activity, through the loss of inhibitory subunits.[5] In this more active state, G proteins can bind other proteins and turn on downstream signalling targets. this whole process is regulated by GAPs, which can down regulate the activity of G proteins.
G proteins can weakly hydrolyse GTP, breaking a phosphate bond to make GDP.[5] In the GDP-bound state, the G proteins are subsequently inactivated and can no longer bind their targets.[5] This hydrolysis reaction, however, occurs very slowly, meaning G proteins have a built-in timer for their activity. G proteins have a window of activity followed by slow hydrolysis, which turns them off. GAP accelerates this G protein timer by increasing the hydrolytic GTPase activity of the G proteins, hence the name GTPase-activating protein.

To be specific, it appears that GAPs serve to make GTP on the G protein a better substrate for nucleophilic attack and lower the transition state energy for the hydrolysis reaction. For example, many GAPs of the small G proteins have a conserved finger-like domain, usually an arginine finger, which changes the conformation of the GTP-bound G protein to orient the GTP for better nucleophilic attack by water.[6] This makes the GTP a better substrate for the reaction. Similarly, GAPs seem to induce a GDP-like charge distribution in the bound GTP.[7] Because the change in charge distribution makes the GTP substrate more like the products of the reaction, GDP and monophoshate, this, along with opening the molecule for nucleophilic attack, lowers the transition state energy barrier of the reaction and allows GTP to be hydrolyzed more readily. GAPs, then, work to enhance the GTP hydrolysis reaction of the G proteins. By doing so, they accelerate the G protein’s built-in timer, which inactivates the G proteins more quickly, and along with the inactivation of GEFs, this keeps the G protein signal off. GAPs, then, are critical in the regulation of G proteins.
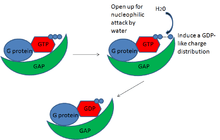
GAP specificity to G proteins
In general, GAPs tend to be pretty specific for their target G proteins. The exact mechanism of target specificity is not fully known, but it is likely that this specificity comes from a variety of factors. At the most basic level, GAP-to-G protein specificity may come simply from the timing and location of protein expression. RGS9-1, for example, is specifically expressed in the rod and cone photoreceptors in the eye retina, and is the only one to interact with G proteins involved in phototransduction in this area.[8] A certain GAP and a certain G protein happen to be expressed in the same time and place, and that is how the cell ensures specificity. Meanwhile, scaffold proteins can also sequester the proper GAP to its G protein and enhance the proper binding interactions.[8] These binding interactions may be specific for a particular GAP and G protein. Also, GAPs may have particular amino acid domains that recognize only a particular G protein. Binding to other G proteins may not have the same favorable interactions, and they therefore do not interact. GAPs can, therefore, regulate specific G proteins.
Examples and classification
EIF5 is a GTPase-activating protein.[9] Furthermore, YopE is a protein domain that is a Rho GTPase-activating protein (GAP), which targets small GTPases such as RhoA, Rac1, and Rac2.[10]
Monomeric
The GAPs that act on small GTP-binding proteins of the Ras superfamily have conserved structures and use similar mechanisms,
An example of a GTPase is the monomer Ran, which is found in the cytosol as well as the nucleus. Hydrolysis of GTP by Ran is thought to provide the energy needed to transport nuclear proteins into the cell. Ran is turned on and off by GEFs and GAPs, respectively.
Heterotrimeric
Most GAPs that act on alpha subunits of heterotrimeric G proteins belong to a distinct family, the RGS protein family.
Regulation of GAPs
While GAPs serve to regulate the G proteins, there is also some level of regulation of the GAPs themselves. Many GAPs have allosteric sites that serve as interfaces with downstream targets of the particular path that they regulate. For example, RGS9-1, the GAP in the photoreceptors from above, interacts with cGMP phosphodiesterase (cGMP PDE), a downstream component of phototransduction in the retina. Upon binding with cGMP PDE, RGS9-1 GAP activity is enhanced.[8] In other words, a downstream target of photoreceptor-induced signaling binds and activates the inhibitor of signaling, GAP. This positive regulatory binding of downstream targets to GAP serves as a negative feedback loop that eventually turns off the signaling that was originally activated. GAPs are regulated by targets of the G protein that they regulate.
There are also examples of negative regulatory mechanisms, where downstream targets of G protein signaling inhibit the GAPs. In G protein-gated potassium channels, phosphatidylinositol 3, 4, 5-triphosphate (PIP3) is a downstream target of G protein signaling. PIP3 binds and inhibits the RGS4 GAP.[11] Such inhibition of GAP may perhaps “prime” the signaling pathway for activation. This creates a window of activity for the G proteins once activated because the GAP is temporarily inhibited. When the potassium channel is activated, Ca2+ gets released and binds calmodulin. Together, they displace PIP3 from GAP by binding competitively to the same site, and by doing so, they reactivate GAP to turn G protein signaling off.[11] This particular process demonstrates both inhibition and activation of GAP by its regulators. There is cross-talk between GAP and other components of the signaling pathway that regulate the activity of GAP.
It is interesting to note that there have been some findings suggesting the possibility of crosstalk between GAPs. A recent study showed that the p120Ras GAP could bind the DLC1 Rho GAP at its catalytic domain. The binding of the Ras GAP to the Rho GAP inhibits the activity of the Rho GAP, thereby activating the Rho G protein.[12] One GAP serves as a negative regulator of another GAP. The reasons for such cross-regulation across GAPs are yet unclear, but one possible hypothesis is that this cross-talk across GAPs attenuates the “off” signal of all the GAPs. Although the p120Ras GAP is active, therefore inhibiting that particular pathway, other cellular processes can still continue because it inhibits other GAPs. This may ensure that the whole system does not shut down from a single off signal. GAP activity is highly dynamic, interacting with many other components of signaling pathways.
Disease associations and clinical relevance
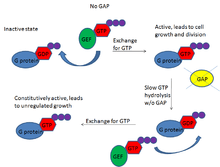
The importance of GAPs comes from its regulation of the crucial G proteins. Many of these G proteins are involved in cell cycling, and as such are known proto-oncogenes. The Ras superfamily of G proteins, for example, has been associated with many cancers because Ras is a common downstream target of many growth factors like FGF, or fibroblast growth factor.[13] Under normal conditions, this signaling ultimately induces regulated cell growth and proliferation. However, in the cancer state, such growth is no longer regulated and results in the formation of tumors.

Often, this oncogenic behavior is due to a loss of function of GAPs associated with those G proteins or a loss of the G protein’s ability to respond to its GAP. With the former, G proteins are unable to hydrolyze GTP quickly, resulting in sustained expression of the active form of G proteins. Although the G proteins have weak hydrolytic activity, in the presence of functional GEFs, the inactivated G proteins are constantly replaced with activated ones because the GEFs exchange GDP for GTP in these proteins. With no GAPs to curb the G protein’s activity, this results in constitutively active G proteins, unregulated cell growth, and the cancerous state. In the case of the latter, a loss of the G protein’s ability to respond to GAP, the G proteins have lost their ability to hydrolyze GTP. With a nonfunctional G protein enzyme, GAPs cannot activate the GTPase activity, and the G protein is constitutively on. This also results in unregulated cell growth and cancer.
Examples of GAP malfunction are ubiquitous clinically. Some cases involve a decreased expression of the GAP gene. For example, some recently characterized cases of papillary thyroid cancer cells in patients show a decreased expression of Rap1GAP, and this expression is seemingly caused by a decreased expression of the GAP mRNA, shown by qRT-PCR experiments.[14] In this case, there appears to be a loss of proper Rap1GAP gene expression. In another case, expression of the Ras GAP is lost in several cancers due to improper epigenetic silencing of the gene. These cells have CpG methylations near the gene that, in effect, silence gene transcription.[15] Regulation of G proteins is lost because the regulator is absent, resulting in cancer.
Other cancers show a loss of sensitivity of the G protein to the GAPs. These G proteins acquire missense mutations that disrupt the inherent GTPase activity of the proteins. The mutant G proteins are still bound by GAPs,[16] but enhancing GTPase activity by the GAPs is meaningless when GTPase activity of the G protein itself is lost. GAP works to activate a nonfunctional hydrolytic enzyme. T24 bladder cancer cells, for example, were shown to have a missense mutation, V12G, resulting in constitutively active Ras protein.[17] Despite the presence of the G protein regulator, regulation is lost due to a loss of function in the G protein itself. This loss of function also manifests itself in cancer. GAPs and their interaction with G proteins are, therefore, highly important clinically and are potential targets for cancer therapies.

References
- ↑ Gerhard Krauss (2008). Biochemistry of signal transduction and regulation. Wiley-VCH. pp. 235–. ISBN 978-3-527-31397-6. Retrieved 15 December 2010.
- 1 2 Kimple, A.J. “Structural Determinants of G-protein α Subunit Selectivity by Regulator of G-protein Signaling 2 (RGS2)”. The Journal of Biological Chemistry. 284 (2009): 19402-19411.
- ↑ Xu, Haiming et al. “Loss of the Rho GTPase Activating Protein p190-B Enhances Hematopoietic Stem Cell Engraftment Potential”. Blood. 114 (2009): 3557-3566.
- ↑ Krendel, M. “Nucleotide Exchange Factor GEF-H1 Mediates Cross-Talk between Microtubules and the Actin Cytoskeleton”. Nature Cell Biology. 4 (2002): 294-301.
- 1 2 3 Berg et al. “Signal-Transduction Pathways”. Biochemistry. New York: W.H. Freeman and Company, 2007.
- ↑ Scheffzek, K. et al. “The Ras-RasGAP Complex: Structural Basis for GTPase Activation and Its Loss in Oncogenic Ras Mutants”. Science. 277 (1997): 333-338.
- ↑ Kötting, C. et al. “Time-Resolved FTIR Studies Provide Activation Free Energy, Activation Enthalpy and Activation Entropy for GTPase Reactions”. Chemical Physics. 307 (2004): 227-232.
- 1 2 3 Xie, Guo-xi et al. “How Regulators of G Protein Signaling Achieve Selective Regulation”. Journal of Molecuar Biology. 366 (2007): 349-365.
- ↑ Das S, Ghosh R, Maitra U (March 2001). "Eukaryotic translation initiation factor 5 functions as a GTPase-activating protein". J. Biol. Chem. 276 (9): 6720–6. doi:10.1074/jbc.M008863200. PMID 11092890.
- ↑ Rosqvist R, Forsberg A, Rimpiläinen M, Bergman T, Wolf-Watz H (April 1990). "The cytotoxic protein YopE of Yersinia obstructs the primary host defence". Mol. Microbiol. 4 (4): 657–67. doi:10.1111/j.1365-2958.1990.tb00635.x. PMID 2191183.
- 1 2 Ishii, Masaru et al. “Phosphatidylinositol 3,4,5-triphosphate and Ca2+/Calmodulin Competitively Bind to the Regulators of G-Protein-Signaling (RGS) Domain of RGS4 and Reciprocally Regulate Its Action”. Biochemistry Journal. 385 (2005): 65-73.
- ↑ Yang, Xu-Yu et al. “p120Ras-GAP Binds the DLC1 Rho-GAP Tumor Suppressor Protein and Inhibits Its RhoA GTPase and Growth Suppressing Activities”. Oncogene. 28 (2009): 1401-1409.
- ↑ Berg et al. “Signal-Transduction Pathways”. Biochemistry. New York: W.H. Freeman and Company, 2007.
- ↑ Nellore, Anoma et al. “Loss of Rap1GAP in Papillary Thyroid Cancer”. Journal of Clinical Endocrinology and Metabolism. 94 (2009): 1026-1032.
- ↑ Jin, Hongchuan et al. “Epigenetic Silencing of a Ca2+-Regulated Ras GTPase-Activating Protein RASAL Defines a New Mechanism of Ras Activation in Human Cancers”. Proceedings of the National Academy of Sciences of the United States of America. 104 (2007): 12353-12358.
- ↑ Raepple, D. et al. “Determination of Ras-GTP and Ras-GDP in Patients with Acute Myelogenous Leukemia (AML), Myeloproliferative Syndrome (MPS), Juvenile Myelomonocytic Leukemia (JMML), Acute Lymphocytic Leukemia (ALL), and Malignant Lymphoma: Assessment of Mutational and Indirect Activation”. Annals of Hematology. 88 (2009): 319-324.
- ↑ Premkumar Reddy, E. et al. “A Point Mutation is Responsible for the Acquisition of Transforming Properties by the T24 Human Bladder Carcinoma Oncogene”. Nature. 300 (1982): 149-152.
External links
- GTPase-Activating Proteins at the US National Library of Medicine Medical Subject Headings (MeSH)