Dermatopathia pigmentosa reticularis
| Dermatopathia pigmentosa reticularis | |
|---|---|
| Classification and external resources | |
| Specialty | medical genetics |
| ICD-10 | Q82.4 |
| OMIM | 125595 |
Dermatopathia pigmentosa reticularis (DPR), also known as dermatopathia pigmentosa reticularis hyperkeratotica et mutilans, dermatopathia pigmentosa reticularis hypohidotica et atrophica and dermatopathic pigmentosa reticularis,[1]:511 is a rare, autosomal dominant[2] congenital disorder that is a form of ectodermal dysplasia. Dermatopathia pigmentosa reticularis is composed of the triad of generalized reticulate hyperpigmentation, noncicatricial alopecia, and onychodystrophy.[3]:856
Presentation
Symptoms include lack of sweat glands, thin hair, brittle nails, mottled skin, and lack of fingerprints.
Cause
DPR is comparable to Naegeli syndrome, both of which are caused by a specific defect in the keratin 14 protein.[4]
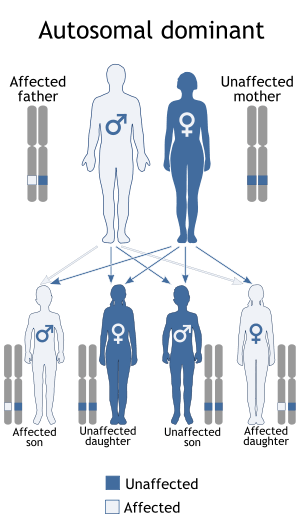
Dermatopathia pigmentosa reticularis has an autosomal dominant pattern of inheritance.
See also
- Naegeli–Franceschetti–Jadassohn syndrome
- List of cutaneous conditions
- List of cutaneous conditions caused by mutations in keratins
References
- ↑ Freedberg, et al. (2003). Fitzpatrick's Dermatology in General Medicine. (6th ed.). McGraw-Hill. ISBN 0-07-138076-0.
- ↑ Heimer WL II, Brauner G, James WD (1992). "Dermatopathia pigmentosa reticularis: a report of a family demonstrating autosomal dominant inheritance". J Am Acad Dermatol. 6 (2 pt. 2): 298–301. doi:10.1016/0190-9622(92)70039-I. PMID 1303619.
- ↑ James, William; Berger, Timothy; Elston, Dirk (2005). Andrews' Diseases of the Skin: Clinical Dermatology. (10th ed.). Saunders. ISBN 0-7216-2921-0.
- ↑ Lugassy J, Itin P, Ishida-Yamamoto A, et al. (October 2006). "Naegeli-Franceschetti-Jadassohn syndrome and dermatopathia pigmentosa reticularis: two allelic ectodermal dysplasias caused by dominant mutations in KRT14". Am. J. Hum. Genet. 79 (4): 724–30. doi:10.1086/507792. PMC 1592572
 . PMID 16960809.
. PMID 16960809.
External links
This article is issued from Wikipedia - version of the 5/26/2016. The text is available under the Creative Commons Attribution/Share Alike but additional terms may apply for the media files.