11-Deoxycorticosterone
 | |
| Names | |
|---|---|
| IUPAC name
(1S,2R,10S,11S,14S,15S)-14-(2-hydroxyacetyl)-2,15-dimethyltetracyclo[8.7.0.02,7.011,15]heptadec-6-en-5-one | |
| Other names
Deoxycorticosterone; Desoxycortone; Deoxycortone; Cortexone; 21-Hydroxyprogesterone; 21-Hydroxy-4-pregnene-3,20-dione; Reichstein's substance Q; Kendall's desoxy compound B; NSC-11319 | |
| Identifiers | |
| 64-85-7 | |
| 3D model (Jmol) | Interactive image |
| ChEBI | CHEBI:16973 |
| ChEMBL | ChEMBL1498 |
| ChemSpider | 5932 |
| ECHA InfoCard | 100.000.543 |
| 2871 | |
| PubChem | 6166 |
| UNII | 40GP35YQ49 |
| |
| |
| Properties | |
| C21H30O3 | |
| Molar mass | 330.461 g/mol |
| Pharmacology | |
| H02AA03 (WHO) | |
| Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa). | |
| Infobox references | |
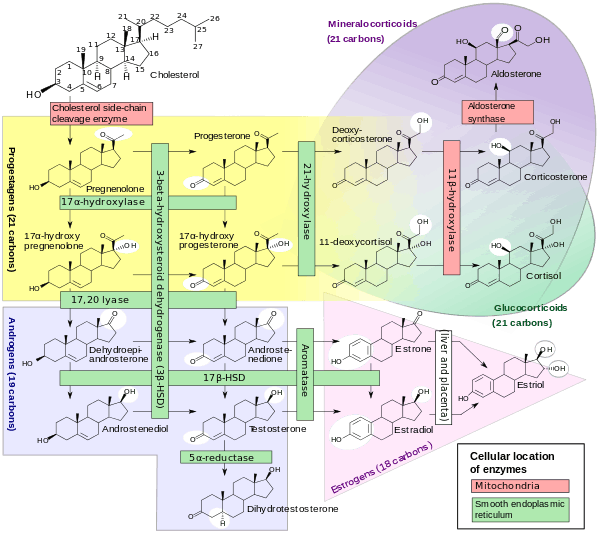
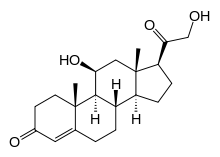
11-Deoxycorticosterone (DOC), or simply deoxycorticosterone, also known as 21-hydroxyprogesterone, as well as desoxycortone (INN), deoxycortone (BAN), and cortexone,[1][2] is a steroid hormone produced by the adrenal gland that possesses mineralocorticoid activity and acts as a precursor to aldosterone.[3] It is an active (Na+-retaining) mineralocorticoid.[4] As its names indicate, 11-deoxycorticosterone can be understood as the 21-hydroxy- variant of progesterone or as the 11-deoxy- variant of corticosterone.
Biological activity
DOC is a potent mineralocorticoid but is virtually devoid of glucocorticoid activity.[5] However, 11-hydroxylation of DOC produces corticosterone and confers glucocorticoid activity, along with 10-fold reduced mineralocorticoid activity.[5] In addition to its mineralocorticoid activity, DOC has been found to possess one-third to one-tenth the potency of progesterone as a progestogen when administered systematically to rabbits.[6] However, it has no such activity when applied directly to the uterine mucosa of mice.[6] The discrepancy may be related to the fact that DOC can be converted into progesterone in vivo.[6]
Biological role
DOC is a precursor molecule for the production of aldosterone. The major pathway for aldosterone production is in the adrenal glomerulosa zone of the adrenal gland. It is not a major secretory hormone. It is produced from progesterone by 21β-hydroxylase and is converted to corticosterone by 11β-hydroxylase. Corticosterone is then converted to aldosterone by aldosterone synthase.[7]
Most of the DOC is secreted by the zona fasciculata of the adrenal cortex which also secretes cortisol, and a small amount by the zona glomerulosa, which secretes aldosterone. DOC stimulates the collecting tubules (the tubules which branch together to feed the bladder) [8] to continue to excrete potassium in much the same way that aldosterone does but not like aldosterone in the end of the looped tubules (distal).[9] At the same time it is not nearly so rigorous at retaining sodium as aldosterone,[10] more than 20 times less. DOC accounts for only 1% of the sodium retention normally [11] In addition to its inherent lack of vigor there is an escape mechanism controlled by an unknown non steroid hormone [12] which overrides DOC's sodium conserving power after a few days just as aldosterone is overridden also.[13] This hormone may be the peptide hormone kallikrein,[14] which is augmented by DOC and suppressed by aldosterone.[15] If sodium becomes very high, DOC also increases urine flow.[8] DOC has about 1/20 of the sodium retaining power of aldosterone,[16] and is said to be as little as one per cent of aldosterone at high water intakes.[17] Since DOC has about 1/5 the potassium excreting power of aldosterone,[16] it probably must have aldosterone's help if the serum potassium content becomes too high. DOC's injections do not cause much additional potassium excretion when sodium intake is low.[18] This is probably because aldosterone is already stimulating potassium outflow. When sodium is low DOC probably would not have to be present, but when sodium rises aldosterone declines considerably, and DOC probably tends to take over.
DOC has a similar feedback with respect to potassium as aldosterone. A rise in serum potassium causes a rise in DOC secretion.[19] However, sodium has little effect,[20] and what effect it does have is direct.[16] Angiotensin (the blood pressure hormone) has little effect on DOC,[19] but DOC causes a rapid fall in renin, and therefore angiotensin I, the precursor of angiotensin II.[21] Therefore, DOC must be indirectly inhibiting aldosterone since aldosterone depends on angiotensin II. Sodium, and therefore blood volume, is difficult to regulate internally. That is, when a large dose of sodium threatens the body with high blood pressure, it cannot be resolved by transferring sodium to the intracellular (inside the cell) space. The red cells would have been possible, but that would not change the blood volume. Potassium, on the other hand, can be moved into the large intracellular space, and apparently it is by DOC in rabbits.[21] Thus, a problem in high blood potassium can be resolved somewhat without jettisoning too much of what is sometimes a dangerously scarce mineral that can not be pumped actively independently from sodium. It is imperative to keep total potassium adequate because a deficiency causes the heart to lose force.[22] Movement of potassium into the cells would intensify the sodium problem somewhat because when potassium moves into the cell, a somewhat smaller amount of sodium moves out.[23] Thus, it is desirable to resolve the blood pressure problem as much as possible by the fall in renin above, therefore avoiding loss of sodium, which was usually in very short supply on the African savannas where human ancestors probably evolved.
The resemblance of the pattern of the electromotive forces produced by DOC in the kidney tubules to normal potassium intake, and the total dissimilarity of their shape as produced by potassium deficient tubules,[24] would tend to support the above view. The above attributes are consistent with a hormone which is relied upon to unload both excess sodium and potassium. DOC's action in augmenting kallikrein, the peptide hormone thought to be the sodium "escape hormone," and aldosterone's action in suppressing it,[15] is also supportive of the above concept.
ACTH has more effect on DOC than it does on aldosterone. This may be to give the immune system control over the electrolyte regulation during diarrhea [25] since during dehydration, aldosterone virtually disappears [26] any way even though renin and angiotensin rise high. It is for this aldosterone disappearance reason that potassium supplements are very dangerous during dehydration and must not be attempted until at least one hour after rehydration in order to give time for the hormones to reach the nucleus.
DOC's primary purpose is to regulate electrolytes. However, it has other effects, such as to remove potassium from leucocytes [27] and muscle,[28] depress glycogen formation [29] and to stimulate copper containing lysyl oxidase enzyme [30] and connective tissue,[31] which attributes may be used by the body to help survive during potassium wasting intestinal diseases.[25] The greater efficiency of DOC in permitting sodium excretion (or perhaps it should be expressed as inefficiency at retention) must be partly through morphological changes in the kidney cells because escape from DOC's sodium retention takes several days to materialize, and when it does, these cells are much more efficient at unloading sodium if sodium is then added than cells accustomed to a prior low intake. Thus, paradoxically, a low salt intake should be protective against loss of sodium in perspiration.
Progesterone prevents some of the loss of potassium by DOC.[32]
Synthesis
The sequence for preparing the hydroxyketone started by conversion of the acid (1) to its chloride with thionyl chloride. Reaction of that acid halide with diazomethane gives the diazoketone (). The hydroxyl group at C3 is then oxidized to the corresponding ketone by means of an Oppenauer reaction. Treatment of the product () with gaseous hydrogen chloride replaces nitrogen in that intermediate by chlorine. Displacement of chlorine by acetate then leads to the 21-acetate (). Saponification of the ester completes the sequence.
Additional images
See also
References
- ↑ Buckingham, MacDonald & Heilbron 1995.
- ↑ Swiss Pharmaceutical Society 2011.
- ↑ Costanzo 2007.
- ↑ Harper's Illustrated Biochemistry 30th Edition
- 1 2 H. Maurice Goodman (28 July 2010). Basic Medical Endocrinology. Academic Press. pp. 64–. ISBN 978-0-08-092055-9.
- 1 2 3 The Adrenocortical Hormones: Their Origin · Chemistry, Physiology, and Pharmacology. Springer Science & Business Media. 27 November 2013. pp. 610–. ISBN 978-3-642-88385-9.
- ↑ Lieberman, Marks & Peet 2012.
- 1 2 O'Neil & Helmans 1977.
- ↑ Peterson & Wright 1977.
- ↑ Ellinghaus 1971.
- ↑ Ruch & Fulton 1960.
- ↑ Pearce et al. 1969.
- ↑ Schacht, Lowenstein & Baldwin 1971.
- ↑ Majima et al. 1999.
- 1 2 Bönner et al. 1981.
- 1 2 3 Oddie, Coghlan & Scoggins 1972.
- ↑ Desaulles 1958.
- ↑ Bauer & Gaunter 1979.
- 1 2 Brown, Strott & Liddle 1972.
- ↑ Schambelan & Biglier 1972.
- 1 2 Grekin, Terris & Bohr 1980.
- ↑ Abbrecht 1972.
- ↑ Rubini & Chojnocki 1972.
- ↑ O'Neil & Helman 1977.
- 1 2 Weber 1998.
- ↑ Merrill, Skelton & Cowley 1986.
- ↑ Wilson 1957.
- ↑ Tobian & Binion 1954.
- ↑ Bartlett & MacKay 1949.
- ↑ Weber 1984.
- ↑ Pospísilová & Pospísil 1970.
- ↑ Wambach & Higgins 1979.
Sources
- Abbrecht, P. H. (1972). "Cardiovascular effects of chronic potassium deficiency in the dog". American Journal of Physiology. 223 (3): 555–560. PMID 4403425.
- Bartlett, G. R.; MacKay, E. M. (1949). "Insulin stimulation of glycogen formation in rat abdominal muscle". Proceedings of The Society for Experimental Biology and Medicine. 71 (3): 493–495. doi:10.3181/00379727-71-17234. PMID 18136520.
- Bauer, J. H.; Gauntner, W. C. (1979). "Effect of potassium chloride on plasma renin activity and plasma aldosterone during sodium restriction in normal man". Kidney International. 15: 286–293. doi:10.1038/ki.1979.37. PMID 513492.
- Bönner, G.; Autenreith, R.; Marin-Grez, M.; Rascher, W.; Gross, F. (1981). "Effects of Sodium Loading, Desoxycorticosterone Acetate, and Corticosterone on Urinary Kallikrein Excretion". Horm. Res. 14 (2): 87–94. doi:10.1159/000179365.
- Brown, R. D.; Strott, C. A.; Liddle, G. W. (1972). "Site of stimulation of aldosterone biosynthesis by angiotensin and potassium". Journal of Clinical Investigation. 51 (6): 1413–1418. doi:10.1172/JCI106937. PMC 292278
 . PMID 4336939.
. PMID 4336939.
- Buckingham, J.; MacDonald, F.; Heilbron, Sir I. (eds.). Dictionary of Organic Compounds. 1 (6th ed.). CRC Press. ISBN 978-0412540905.
- Costanzo, Linda S. (2014). Physiology. Board Review Series (BRS) (International ed.). Lippincott Williams & Wilkins. ISBN 978-1469832005.
- Desaulles, P. (1958). Muller, A. F.; O'Connor, C. M., eds. Comparison of the effects of aldosterone, cortexone and cortisol on adrenalectomized rats under various salt loads. An International Symposium on Aldosterone, Geneva 1957. Boston: Little Brown Co. pp. 29–38. OCLC 14594852.
- Ellinghaus, K. (1971). "Sodium and potassium balance during the administration of desoxycorticosterone in dogs with differing intakes". Pflügers Archiv. 322 (4): 347–354. doi:10.1007/BF00587752.
- Grekin, R.J.; Terris, J. M.; Bohr, D. F. (1980). "Electrolyte and hormonal effects of deoxycorticosterone acetate in young pigs". Hypertension. 2 (3): 326–332. doi:10.1161/01.HYP.2.3.326. PMID 6993359.
- Lieberman, Michael A.; Marks, Allan; Peet, Alisa. Marks' Basic Medical Biochemistry: A Clinical Approach. Matthew Chansky (Illustrator) (4th ed.). Lippincott Williams and Wilkins. ISBN 978-1608315727.
- Majima, M.; Hayashi, I.; Fujita, T.; Ito, H.; Nakajima, S.; Katori, M. (1999). "Facilitation of renal kallikrein-kinin system prevents the development of hypertension by inhibition of sodium retention". Immunopharmacology. 44 (1-2): 145–152. doi:10.1016/S0162-3109(99)00086-7. PMID 10604538.
- Merrill, D. C.; Skelton, M. M.; Cowley, A. W. (1986). "Humoral control of water and electrolyte excretion during water restriction". Kidney International. 29 (6): 1152–1161. doi:10.1038/ki.1986.121.
- Oddie, C. J.; Coghlan, J. P.; Scoggins, B. (1972). "Plasma Deoxycorticosterone Levels in Man with Simultaneous Measurement of Aldosterone, Corticosterone, Cortisol and 11-Deoxycortisol". Journal of Clinical Endocrinology & Metabolism. 34 (6): 1039–1054. doi:10.1210/jcem-34-6-1039. PMID 4336456.
- O'Neil, R. G.; Helmans, S. I. (1977). "Transport characteristics of renal collecting tubules: influence of DOCA and diet". American Journal of Physiology. 233 (6): F544–558. PMID 596453.
- Pearce, J. W.; Sonnenberg, H.; Veress, A. T.; Ackermann, U. (1969). "Evidence for a humoral factor modifying the renal response to blood volume expansion in the rat". Canadian Journal of Physiology and Pharmacology. 47 (4): 377–386. doi:10.1139/y69-066.
- Peterson, L.; Wright, F. S. (1977). "Effect of sodium intake on renal potassium excretion". American Journal of Physiology. 233 (3): 225–234. PMID 910918.
- Pospísilová, J.; Pospísil, M. (1970). "Influence of mineralocorticoids on collagen synthesis in subcutaneous granuloma in adrenalectomized and nonadrenalectomized mice". Physiologia Bohemoslovaca. 19 (6): 539–543. PMID 4251388.
- Rubini, M. E.; Chojnocki, R. F. (1972). "Principles of parenteral therapy". Am J Clin Nutr. 25 (1): 96–113. PMID 5007603.
- Ruch, T. C.; Fulton, J. F., eds. (1960). Medical Physiology and Biophysics. W. B. Saunders and Co.
- Schacht, R. G.; Lowenstein, J.; Baldwin, D. S. (1971). "Renal mechanism for DOCA escape in man". Bulletin of New York Academy of Medicine. 47 (10): 1233. PMID 5286466.
- Schambelan, M.; Biglieri, E. C. (1972). "Deoxycorticosterone production and regulation in man". Journal of Clinical Endocrinology and Metabolism. 34 (4): 695–703. doi:10.1210/jcem-34-4-695.
- Swiss Pharmaceutical Society (2011). Index Nominum: International Drug Directory (20th ed.). Wissenschaftliche Verlag. ISBN 978-3804750531.
- Tobian, L.; Binion, J. T. (1954). "Artery Wall Electrolytes in Renal and DCA Hypertension". Journal of Clinical Investigation. 23 (10): 1407–1414. doi:10.1172/JCI103018.
- Wambach, G; Higgins, J. R. (1979). "Effect of progesterone on serum and tissue electrolyte concentration in DOCA-treated rats". Hormone Metabolism Research. 11 (3): 258–259. doi:10.1055/s-0028-1095777. PMID 447212.
- Weber, C. E. (1984). "Copper response to rheumatoid arthritis". Medical Hypotheses. 15 (4): 333–348. doi:10.1016/0306-9877(84)90150-6. PMID 6152006.
- Weber, C. E. (1998). "Cortisol's purpose". Medical Hypotheses. 51 (4): 289–292. doi:10.1016/S0306-9877(98)90049-4. PMID 9824832.
- Wilson, D. L. (1957). "Direct effects of adrenal corticosteroids on the electrolyte content of rabbit leucocytes". American Journal of Physiology. 190 (1): 104–108. PMID 13458419.