Cladogram

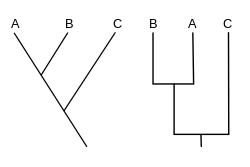
A cladogram (from Greek clados "branch" and gramma "character") is a diagram used in cladistics which shows relations among organisms. A cladogram is not, however, an evolutionary tree because it does not show how ancestors are related to descendants or how much they have changed; many evolutionary trees can be inferred from a single cladogram.[1][2][3][4][5] A cladogram uses lines that branch off in different directions ending at a clade, a groups of organisms with a last common ancestor. There are many shapes of cladograms but they all have lines that branch off from other lines. The lines can be traced back to where they branch off. These branching off points represent a hypothetical ancestor (not an actual entity) which is inferred to exhibit the traits shared among the terminal taxa above it.[4][6] This hypothetical ancestor might then provide clues about the order of evolution of various features, adaptation, and other evolutionary narratives about ancestors. Although traditionally such cladograms were generated largely on the basis of morphological characters, DNA and RNA sequencing data and computational phylogenetics are now very commonly used in the generation of cladograms, either on their own or in combination with morphology.
Generating a cladogram
Molecular versus morphological data
The characteristics used to create a cladogram can be roughly categorized as either morphological (synapsid skull, warm blooded, notochord, unicellular, etc.) or molecular (DNA, RNA, or other genetic information).[7] Prior to the advent of DNA sequencing, cladistic analysis primarily used morphological data. Behavioral data (for animals) may also be used.[8]
As DNA sequencing has become cheaper and easier, molecular systematics has become a more and more popular way to infer phylogenetic hypotheses.[9] Using a parsimony criterion is only one of several methods to infer a phylogeny from molecular data; maximum likelihood and Bayesian inference, which incorporate explicit models of sequence evolution, are non-Hennigian ways to evaluate sequence data. Another powerful method of reconstructing phylogenies is the use of genomic retrotransposon markers, which are thought to be less prone to the problem of reversion that plagues sequence data. They are also generally assumed to have a low incidence of homoplasies because it was once thought that their integration into the genome was entirely random; this seems at least sometimes not to be the case, however.

Plesiomorphies and synapomorphies
Researchers must decide which character states are "ancestral" (plesiomorphies) and which are derived (synapomorphies), because only synapomorphic character states provide evidence of grouping.[10] This determination is usually done by comparison to the character states of one or more outgroups. States shared between the outgroup and some members of the in-group are symplesiomorphies; states that are present only in a subset of the in-group are synapomorphies. Note that character states unique to a single terminal (autapomorphies) do not provide evidence of grouping. The choice of an outgroup is a crucial step in cladistic analysis because different outgroups can produce trees with profoundly different topologies.
Homoplasies
A homoplasy is a character state that is shared by two or more taxa due to some cause other than common ancestry.[11] The two main types of homoplasy are convergence (evolution of the "same" character in at least two distinct lineages) and reversion (the return to an ancestral character state). Characters that are obviously homoplastic, such as white fur in different lineages of Arctic mammals, should not be included as a character in a phylogenetic analysis as they do not contribute anything to our understanding of relationships. However, homoplasy is often not evident from inspection of the character itself (as in DNA sequence, for example), and is then detected by its incongruence (unparsimonious distribution) on a most-parsimonious cladogram. Note that characters that are homoplastic may still contain phylogenetic signal.
A well-known example of homoplasy due to convergent evolution would be the character, "presence of wings". Though the wings of birds, bats, and insects serve the same function, each evolved independently, as can be seen by their anatomy. If a bird, bat, and a winged insect were scored for the character, "presence of wings", a homoplasy would be introduced into the dataset, and this could potentially confound the analysis, possibly resulting in a false hypothesis of relationships. Of course, the only reason a homoplasy is recognizable in the first place is because there are other characters that imply a pattern of relationships that reveal its homoplastic distribution.
What is a cladogram and what is not a cladogram?
As noted above, a cladogram is the diagrammatic result of a parsimony analysis, which groups taxa on the basis of synapomorphies alone. There are many other phylogenetic algorithms that treat data somewhat differently, and result in phylogenetic trees that look like cladograms but are not cladograms. For example, phenetic algorithms, such as UPGMA and Neighbor-Joining, group by overall similarity, and treat both synapomorphies and symplesiomorphies as evidence of grouping, The resulting diagrams are phenograms, not cladograms, Similarly, the results of model-based methods (Maximum Likelihood or Bayesian approaches) that take into account both branching order and "branch length," count both synapomorphies and autapomorphies as evidence for or against grouping, The diagrams resulting from those sorts of analysis are not cladograms, either.
Cladogram selection
There are several algorithms available to identify the "best" cladogram.[12] Most algorithms use a metric to measure how consistent a candidate cladogram is with the data. Most cladogram algorithms use the mathematical techniques of optimization and minimization.
In general, cladogram generation algorithms must be implemented as computer programs, although some algorithms can be performed manually when the data sets are trivial (for example, just a few species and a couple of characteristics).
Some algorithms are useful only when the characteristic data are molecular (DNA, RNA); other algorithms are useful only when the characteristic data are morphological. Other algorithms can be used when the characteristic data includes both molecular and morphological data.
Algorithms for cladograms include least squares, neighbor-joining, parsimony, maximum likelihood, and Bayesian inference.
Biologists sometimes use the term parsimony for a specific kind of cladogram generation algorithm and sometimes as an umbrella term for all cladogram algorithms.[13]
Algorithms that perform optimization tasks (such as building cladograms) can be sensitive to the order in which the input data (the list of species and their characteristics) is presented. Inputting the data in various orders can cause the same algorithm to produce different "best" cladograms. In these situations, the user should input the data in various orders and compare the results.
Using different algorithms on a single data set can sometimes yield different "best" cladograms, because each algorithm may have a unique definition of what is "best".
Because of the astronomical number of possible cladograms, algorithms cannot guarantee that the solution is the overall best solution. A nonoptimal cladogram will be selected if the program settles on a local minimum rather than the desired global minimum.[14] To help solve this problem, many cladogram algorithms use a simulated annealing approach to increase the likelihood that the selected cladogram is the optimal one.[15]
The basal position is the direction of the base (or root) of a rooted phylogenetic tree or cladogram. A basal clade is the earliest clade (of a given taxonomic rank[a]) to branch within a larger clade.
Cladogram statistics
Incongruence length difference test (or partition homogeneity test)
The incongruence length difference test (ILD) is a measurement of how the combination of different datasets (e.g. morphological and molecular, plastid and nuclear genes) contributes to a longer tree. It is measured by first calculating the total tree length of each partition and summing them. Then replicates are made by making randomly assembled partitions consisting of the original partitions. The lengths are summed. A p value of 0.01 is obtained for 100 replicates if 99 replicates have longer combined tree lengths.
Measuring homoplasy
Some measures attempt to measure the amount of homoplasy in a dataset with reference to a tree,[16] though it is not necessarily clear precisely what property these measures aim to quantify[17]
Consistency index
The consistency index (CI) measures the consistency of a tree to a set of data – a measure of the minimum amount of homoplasy implied by the tree.[18] It is calculated by counting the minimum number of changes in a dataset and dividing it by the actual number of changes needed for the cladogram.[18] A consistency index can also be calculated for an individual character i, denoted ci.
Besides reflecting the amount of homoplasy, the metric also reflects the number of taxa in the dataset,[19] (to a lesser extent) the number of characters in a dataset,[20] the degree to which each character carries phylogenetic information,[21] and the fashion in which additive characters are coded, rendering it unfit for purpose.[22]
ci occupies a range from 1 to 1/[n.taxa/2] in binary characters with an even state distribution; its minimum value is larger when states are not evenly spread.[21][23]
Retention index
The retention index (RI) was proposed as an improvement of the CI "for certain applications"[24] This metric also purports to measure of the amount of homoplasy, but also measures how well synapomorphies explain the tree. It is calculated taking the (maximum number of changes on a tree minus the number of changes on the tree), and dividing by the (maximum number of changes on the tree minus the minimum number of changes in the dataset).
The rescaled consistency index (RC) is obtained by multiplying the CI by the RI; in effect this stretches the range of the CI such that its minimum theoretically attainable value is rescaled to 0, with its maximum remaining at 1.[23][24] The homoplasy index (HI) is simply 1 − CI.
Homoplasy Excess Ratio
This measures the amount of homoplasy observed on a tree relative to the maximum amount of homoplasy that could theoretically be present - 1 − (observed homoplasy excess) / (maximum homoplasy excess).[25] A value of 1 indicates no homoplasy; 0 represents as much homoplasy as there would be in a fully random dataset, and negative values indicate more homoplasy still (and tend only to occur in contrived examples).[25] The HER is presented as the best measure of homoplasy currently available.[23][26]
See also
References
- ↑ Mayr, Ernst (2009). "Cladistic analysis or cladistic classification?". Journal of Zoological Systematics and Evolutionary Research. 12: 94–128. doi:10.1111/j.1439-0469.1974.tb00160.x.
- ↑ Foote, Mike (Spring 1996). "On the Probability of Ancestors in the Fossil Record". Paleobiology. 22 (2): 141–51. JSTOR 2401114.
- ↑ Dayrat, Benoît (Summer 2005). "Ancestor-Descendant Relationships and the Reconstruction of the Tree of Life". Paleobiology. 31 (3): 347–53. doi:10.1666/0094-8373(2005)031[0347:aratro]2.0.co;2. JSTOR 4096939.
- 1 2 Posada, David; Crandall, Keith A. (2001). "Intraspecific gene genealogies: Trees grafting into networks". Trends in Ecology & Evolution. 16: 37–45. doi:10.1016/S0169-5347(00)02026-7.
- ↑ Podani, János (2013). "Tree thinking, time and topology: Comments on the interpretation of tree diagrams in evolutionary/phylogenetic systematics". Cladistics. 29 (3): 315–327. doi:10.1111/j.1096-0031.2012.00423.x.
- ↑ Schuh, Randall T. (2000). Biological Systematics: Principles and Applications. ISBN 978-0-8014-3675-8.
- ↑ DeSalle, Rob (2002). Techniques in Molecular Systematics and Evolution. Birkhauser. ISBN 3-7643-6257-X.
- ↑ Wenzel, John W. (1992). "Behavioral homology and phylogeny". Annu. Rev. Ecol. Syst. 23: 361–381. doi:10.1146/annurev.es.23.110192.002045.
- ↑ Hillis, David (1996). Molecular Systematics. Sinaur. ISBN 0-87893-282-8.
- ↑ Hennig, Willi (1966). Phylogenetic Systematics. University of Illinois Press.
- ↑ West-Eberhard, Mary Jane (2003). Developmental Plasticity and Evolution. Oxford Univ. Press. pp. 353–376. ISBN 0-19-512235-6.
- ↑ Kitching, Ian (1998). Cladistics: The Theory and Practice of Parsimony Analysis. Oxford University Press. ISBN 0-19-850138-2.
- ↑ Stewart, Caro-Beth (1993). "The powers and pitfalls of parsimony". Nature. 361 (6413): 603–7. Bibcode:1993Natur.361..603S. doi:10.1038/361603a0. PMID 8437621.
- ↑ Foley, Peter (1993). Cladistics: A Practical Course in Systematics. Oxford Univ. Press. p. 66. ISBN 0-19-857766-4.
- ↑ Nixon, Kevin C. (1999). "The Parsimony Ratchet, a New Method for Rapid Parsimony Analysis". Cladistics. 15 (4): 407–414. doi:10.1111/j.1096-0031.1999.tb00277.x.
- ↑ reviewed in Archie, James W. (1996). "Homoplasy": 153–188. doi:10.1016/B978-012618030-5/50008-3. ISBN 9780126180305.
|chapter=ignored (help) - ↑ CHANG, J. T. and KIM, J. 1996. The measurement of homoplasy: a stochastic view. In SANDERSON, M. J. and HUFFORD, L. (eds.) Homoplasy: The Recurrence of Similarity in Evolution, Academic Press, 189–203 pp.
- 1 2 Kluge, A. G.; Farris, J. S. (1969). "Quantitative Phyletics and the Evolution of Anurans". Systematic Zoology. 18: 1–32. doi:10.2307/2412407.
- ↑ Archie, J. W.; Felsenstein, J. (1993). "The Number of Evolutionary Steps on Random and Minimum Length Trees for Random Evolutionary Data". Theoretical Population Biology. 43: 52–79. doi:10.1006/tpbi.1993.1003.
- ↑ Archie, J. W. (1989). "HOMOPLASY EXCESS RATIOS : NEW INDICES FOR MEASURING LEVELS OF HOMOPLASY IN PHYLOGENETIC SYSTEMATICS AND A CRITIQUE OF THE CONSISTENCY INDEX the data sets and phylogenetic hypotheses . evaluating alternative phylogenetic hy- steps present on a particular t.". Systematic Zoology. 38: 253–269. doi:10.2307/2992286.
- 1 2 Hoyal Cuthill, Jennifer F.; Braddy, Simon J.; Donoghue, Philip C. J. (2010). "A formula for maximum possible steps in multistate characters: Isolating matrix parameter effects on measures of evolutionary convergence". Cladistics. 26: 98–102. doi:10.1111/j.1096-0031.2009.00270.x.
- ↑ Sanderson, M. J.; Donoghue, M. J. (1989). "Patterns of variations in levels of homoplasy". Evolution. 43: 1781–1795. doi:10.2307/2409392.
- 1 2 3 ARCHIE, J. W. 1996. Measures of homoplasy. In Homoplasy, Elsevier, 153–188 pp.
- 1 2 Farris, J. S. (1989). "The retention index and the rescaled consistency index". Cladistics. 5: 417–419. doi:10.1111/j.1096-0031.1989.tb00573.x.
- 1 2 Archie, J. W. (1989). "HOMOPLASY EXCESS RATIOS : NEW INDICES FOR MEASURING LEVELS OF HOMOPLASY IN PHYLOGENETIC SYSTEMATICS AND A CRITIQUE OF THE CONSISTENCY INDEX". Systematic Zoology. 38: 253–269. doi:10.2307/2992286.
- ↑ Hoyal Cuthill, Jennifer (2015). "The size of the character state space affects the occurrence and detection of homoplasy: Modelling the probability of incompatibility for unordered phylogenetic characters". Journal of Theoretical Biology. 366: 24–32. doi:10.1016/j.jtbi.2014.10.033. PMID 25451518.
External links
 Media related to Cladograms at Wikimedia Commons
Media related to Cladograms at Wikimedia Commons