Bayesian inference in phylogeny
| Classification | Evolutionary biology |
|---|---|
| Subclassification | Molecular phylogenetics |
| Optimally search criteria | Bayesian inference |
Bayesian inference of phylogeny uses a likelihood function to create a quantity called the posterior probability of trees using a model of evolution, based on some prior probabilities, producing the most likely phylogenetic tree for the given data. The Bayesian approach has become popular due to advances in computing speeds and the integration of Markov chain Monte Carlo (MCMC) algorithms. Bayesian inference has a number of applications in molecular phylogenetics and systematics.
Bayesian inference of phylogeny background and bases
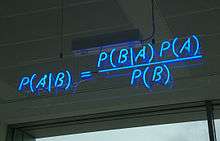

Bayesian inference refers to a probabilistic method developed by Reverend Thomas Bayes based on Bayes' theorem. Published posthumously in 1763 it was the first expression of inverse probability and the basis of Bayesian inference. Independently, unaware of Bayes work, Pierre-Simon Laplace developed Bayes' theorem in 1774.[1]
Bayesian inference was widely used until 1900s when there was a shift to frequentist inference, mainly due to computational limitations. Based on Bayes' theorem, the bayesian approach combines the prior probability of a tree P(A) with the likelihood of the data (B) to produce a posterior probability distribution on trees P(A|B). The posterior probability of a tree will indicate the probability of the tree to be correct, being the tree with the highest posterior probability the one chosen to represent best a phylogeny. It was the introduction of Monte Carlo Markov Chains (MCMC) methods by Nicolas Metropolis in 1953 that revolutionized Bayesian Inference and by the 1990s became a widely used method amongst phylogeneticists. Some of the advantages over traditional maximum parsimony and maximum likelihood methods are the possibility of account for the phylogenetic uncertainty, use of prior information and incorporation of complex models of evolution that limited computational analyses for traditional methods. Although overcoming complex analytical operations the posterior probability still involves a summation over all trees and, for each tree, integration over all possible combinations of substitution model parameter values and branch lengths.
MCMC methods can be described in three steps: first using a stochastic mechanism a new state for the Markov chain is proposed. Secondly, the probability of this new state to be correct is calculated. Thirdly, a new random variable (0,1) is proposed. If this new value is less than the acceptance probability the new state is accepted and the state of the chain is updated. This process is run for either thousands or millions of times. The amount of time a single tree is visited during the course of the chain is just a valid approximation of its posterior probability. Some of the most common algorithms used in MCMC methods include the Metropolis-Hastings algorithms, the Metropolis-Coupling MCMC (MC³) and the LOCAL algorithm of Larget and Simon.
Metropolis-Hastings algorithm
One of the most common MCMC methods used is the Metropolis-Hastings algorithm,[2] a modified version of the original Metropolis algorithm.[3] It is a widely used method to sample randomly from complicated and multi-dimensional distribution probabilities. The Metropolis algorithm is described in the following steps:[4]
- An initial tree, Ti, is randomly selected
- A neighbour tree, Tj, is selected from the collection of trees.
- The ratio, R, of the probabilities (or probability density functions) of Tj and Ti is computed as follows: R = f(Tj)/f(Ti)
- If R ≥ 1, Tj is accepted as the current tree
- If R < 1, Tj is accepted as the current tree with probability R, otherwise Ti is kept
- At this point the process is repeated from Step 2 N times.
The algorithm keeps running until it reaches an equilibrium distribution. It also assumes that the probability of proposing a new tree Tj when we are at the old tree state Ti, is the same probability of proposing Ti when we are at Tj. When this is not the case Hastings corrections are applied. The aim of Metropolis-Hastings algorithm is to produce a collection of states with a determined distribution until the Markov process reaches a stationary distribution. The algorithm has two components:
- A potential transition from one state to another (i → j) using a transition probability function qi,j
- Movement of the chain to state j with probability αi,j and remains in i with probability 1 – αi,j.[5]
Metropolis-coupled MCMC
Metropolis-coupled MCMC algorithm (MC³) [6] has been proposed to solve a practical concern of the Markov chain moving across peaks when the target distribution has multiple local peaks, separated by low valleys, are known to exist in the tree space. This is the case during heuristic tree search under maximum parsimony (MP), maximum likelihood (ML), and minimum evolution (ME) criteria, and the same can be expected for stochastic tree search using MCMC. This problem will result in samples not approximating correctly to the posterior density. The (MC³) improves the mixing of Markov chains in presence of multiple local peaks in the posterior density. It runs multiple (m) chains in parallel, each for n iterations and with different stationary distributions , , where the first one, is the target density, while , are chosen to improve mixing. For example, one can choose incremental heating of the form:





![{\displaystyle \pi _{j}(\theta )=\pi (\theta )^{1/[1+\lambda (j-1)]},\ \ \lambda >0,}](../I/m/2e9d8c159f93b004aa99ba2ce2249578daf6db1e.svg)
so that the first chain is the cold chain with the correct target density, while chains are heated chains. Note that raising the density to the power with has the effect of flattening out the distribution, similar to heating a metal. In such a distribution, it is easier to traverse between peaks (separated by valleys) than in the original distribution. After each iteration, a swap of states between two randomly chosen chains is proposed through a Metropolis-type step. Let be the current state in chain , . A swap between the states of chains and is accepted with probability:








At the end of the run, output from only the cold chain is used, while those from the hot chains are discarded. Heuristically, the hot chains will visit the local peaks rather easily, and swapping states between chains will let the cold chain occasionally jump valleys, leading to better mixing. However, if is unstable, proposed swaps will seldom be accepted. This is the reason for using several chains which differ only incrementally.

An obvious disadvantage of the algorithm is that chains are run and only one chain is used for inference. For this reason, is ideally suited for implementation on parallel machines, since each chain will in general require the same amount of computation per iteration.


LOCAL algorithm of Larget and Simon
The LOCAL algorithms[7] offers a computational advantage over previous methods and demonstrates that a Bayesian approach is able to assess uncertainty computationally practical in larger trees. The LOCAL algorithm is an improvement of the GLOBAL algorithm presented in Mau, Newton and Larget (1999)[8] in which all branch lengths are changed in every cycle. The LOCAL algorithms modifies the tree by selecting an internal branch of the tree at random. The nodes at the ends of this branch are each connected to two other branches. One of each pair is chosen at random. Imagine taking these three selected edges and stringing them like a clothesline from left to right, where the direction (left/right) is also selected at random. The two endpoints of the first branch selected will have a sub-tree hanging like a piece of clothing strung to the line. The algorithm proceeds by multiplying the three selected branches by a common random amount, akin to stretching or shrinking the clothesline. Finally the leftmost of the two hanging sub-trees is disconnected and reattached to the clothesline at a location selected uniformly at random. This would be the candidate tree.
Suppose we began by selecting the internal branch with length that separates taxa and from the rest. Suppose also that we have (randomly) selected branches with lengths and from each side, and that we oriented these branches. Let , be the current length of the clothesline. We select the new length to be , where is a uniform random variable on . Then for the LOCAL algorithm, the acceptance probability can be computed to be:










Assessing convergence
To estimate a branch length of a 2-taxon tree under JC, in which sites are unvaried and are variable, assume exponential prior distribution with rate . The density is . The probabilities of the possible site patterns are:





for unvaried sites, and

Thus the unnormalized posterior distribution is:

or, alternately,

Update branch length by choosing new value uniformly at random from a window of half-width centered at the current value:


where is uniformly distributed between and . The acceptance probability is:



Example: , . We will compare results for two values of , and . In each case, we will begin with an initial length of and update the length times.






Maximum parsimony and maximum likelihood

There are many approaches to reconstructing phylogenetic trees, each with advantages and disadvantages, and there is no straightforward answer to “what is the best method?”. Maximum parsimony (MP) and maximum likelihood (ML) are traditional methods widely used for the estimation of phylogenies and both use character information directly, as Bayesian methods do.
Maximum Parsimony recovers one or more optimal trees based on a matrix of discrete characters for a certain group of taxa and it does not require a model of evolutionary change. MP gives the most simple explanation for a given set of data, reconstructing a phylogenetic tree that includes as few changes across the sequences as possible, this is the one that exhibits the smallest number of evolutionary steps to explain the relationship between taxa. The support of the tree branches is represented by bootstrap percentage. For the same reason that it has been widely used, its simplicity, MP has also received criticism and has been pushed into the background by ML and Bayesian methods. MP presents several problems and limitations. As shown by Felsenstein (1978), MP might be statistically inconsistent,[9] meaning that as more and more data (e.g. sequence length) is accumulated, results can converge on an incorrect tree and lead to long branch attraction, a phylogenetic phenomenon where taxa with long branches (numerous character state changes) tend to appear more closely related in the phylogeny than they really are.
As in maximum parsimony, maximum likelihood will evaluate alternative trees. However it considers the probability of each tree explaining the given data based on a model of evolution. In this case, the tree with the highest probability of explaining the data is chosen over the other ones.[10] In other words, it compares how different trees predict the observed data. The introduction of a model of evolution in ML analyses presents an advantage over MP as the probability of nucleotide substitutions and rates of these substitutions are taken into account, explaining the phylogenetic relationships of taxa in a more realistic way. An important consideration of this method is the branch length, which parsimony ignores, with changes being more likely to happen along long branches than short ones. This approach might eliminate long branch attraction and explain the greater consistency of ML over MP. Although considered by many to be the best approach to inferring phylogenies from a theoretical point of view, ML is computationally intensive and it is almost impossible to explore all trees as there are too many. Bayesian inference also incorporates a model of evolution and the main advantages over MP and ML are that it is computationally more efficient than traditional methods, it quantifies and addresses the source of uncertainty and is able to incorporate complex models of evolution.
Pitfalls and controversies
- Boostrap values vs Posterior Probabilities. It has been observed that bootstrap support values, calculated under parsimony or maximum likelihood, tend to be lower than the posterior probabilities obtained by Bayesian inference.[11] This fact leads to a number of questions such as: Do posterior probabilities lead to overconfidence in the results? Are bootstrap values more robust than posterior probabilities?
- Controversy of using prior probabilities. Using prior probabilities for Bayesian analysis has been seen by many as an advantage as it will provide a hypothesis a more realistic view of the real world. However some biologists argue about the subjectivity of Bayesian posterior probabilities after the incorporation of these priors.
- Model choice. The results of the Bayesian analysis of a phylogeny are directly correlated to the model of evolution chosen so it is important to choose a model that fits the observed data, otherwise inferences in the phylogeny will be erroneous. Many scientists have raised questions about the interpretation of Bayesian inference when the model is unknown or incorrect. For example, an oversimplified model might give higher posterior probabilities[12][13] or simple evolutionary model are associated to less uncertainty than that from bootstrap values.[14]
MRBAYES software
MrBayes is a free software that performs Bayesian inference of phylogeny. Originally written by John P. Huelsenbeck and Frederik Ronquist in 2001.[15] As Bayesian methods increased in popularity MrBayes became one of the software of choice for many molecular phylogeneticists. It is offered for Macintosh, Windows, and UNIX operating systems and it has a command-line interface. The program uses the standard MCMC algorithm as well as the Metropolis coupled MCMC variant. MrBayes reads aligned matrices of sequences (DNA or amino acids) in the standard NEXUS format.[16]
MrBayes uses MCMC to approximate the posterior probabilities of trees.[17] The user can change assumptions of the substitution model, priors and the details of the MC³ analysis. It also allows the user to remove and add taxa and characters to the analysis. The program uses the most standard model of DNA substitution, the 4x4 also called JC69, which assumes that changes across nucleotides occurs with equal probability.[18] It also implements a number of 20x20 models of amino acid substitution, and codon models of DNA substitution. It offers different methods for relaxing the assumption of equal substitutions rates across nucleotide sites.[19] MrBayes is also able to infer ancestral states accommodating uncertainty to the phylogenetic tree and model parameters.
MrBayes 3 [20] was a completely reorganized and restructured version of the original MrBayes. The main novelty was the ability of the software to accommodate heterogeneity of data sets. This new framework allows the user to mix models and take advantages of the efficiency of Bayesian MCMC analysis when dealing with different type of data (e.g. protein, nucleotide, and morphological). It uses the Metropolis-Coupling MCMC by default.
MrBayes 3.2 new version of MrBayes was released in 2012.[21] The new version allows the users to run multiple analyses in parallel. It also provides faster likelihood calculations and allow these calculations to be delegated to graphics processing unites (GPUs). Version 3.2 provides wider outputs options compatible with FigTree and other tree viewers.
List of phylogenetics software
This table includes some of the most common phylogenetic software used for inferring phylogenies under a Bayesian framework. Some of them do not use exclusively Bayesian methods.
| Name | Description | Method | Author | Website link |
|---|---|---|---|---|
| Armadillo Workflow Platform | Workflow platform dedicated to phylogenetic and general bioinformatic analysis | Inference of phylogenetic trees using Distance, Maximum Likelihood, Maximum Parsimony, Bayesian methods and related workflows | E. Lord, M. Leclercq, A. Boc, A.B. Diallo and V. Makarenkov [22] | http://www.bioinfo.uqam.ca/armadillo. |
| Bali-Phy | Simultaneous Bayesian inference of alignment and phylogeny | Bayesian inference, alignment as well as tree search | M.A. Suchard, B. D. Redelings [23] | http://www.bali-phy.org |
| BATWING | Bayesian Analysis of Trees With Internal Node Generation | Bayesian inference, demographic history, population splits | I. J. Wilson, D. Weale, D.Balding [24] | http://www.maths.abdn.ac.uk/˜ijw |
| Bayes Phylogenies | Bayesian inference of trees using Markov Chain Monte Carlo methods | Bayesian inference, multiple models, mixture model (auto-partitioning) | M. Pagel, A. Meade [25] | http://www.evolution.rdg.ac.uk/BayesPhy.html |
| PhyloBayes / PhyloBayes MPI | Bayesian Monte Carlo Markov Chain (MCMC) sampler for phylogenetic reconstruction. | Non-parametric methods for modeling among-site variation in nucleotide or amino-acid propensities. | N. Lartillot, N. Rodrigue, D. Stubbs, J. Richer [26] | http://www.phylobayes.org |
| BEAST | Bayesian Evolutionary Analysis Sampling Trees | Bayesian inference, relaxed molecular clock, demographic history | A. J. Drummond, A. Rambaut & M. A. Suchard [27] | http://beast.bio.ed.ac.uk |
| BUCKy | Bayesian concordance of gene trees | Bayesian concordance using modified greedy consensus of unrooted quartets | C. Ané, B. Larget, D.A. Baum, S.D. Smith, A. Rokas and B. Larget, S.K. Kotha, C.N. Dewey, C. Ané [28] | http://www.stat.wisc.edu/~ane/bucky/ |
| Geneious (MrBayes plugin) | Geneious provides genome and proteome research tools | Neighbor-joining, UPGMA, MrBayes plugin, PHYML plugin, RAxML plugin, FastTree plugin, GARLi plugin, PAUP* Plugin | A. J. Drummond,M.Suchard,V.Lefort et al. | http://www.geneious.com |
| TOPALi | Phylogenetic inference | Phylogenetic model selection, Bayesian analysis and Maximum Likelihood phylogenetic tree estimation, detection of sites under positive selection, and recombination breakpoint location analysis | I.Milne, D.Lindner, et al.[29] | http://www.topali.org |
Applications
Bayesian Inference has extensively been used by molecular phylogeneticists for a wide number of applications. Some of these include:

- Inference of phylogenies.[30][31]
- Inference and evaluation of uncertainty of phylogenies.[32]
- Inference of ancestral character state evolution.[33][34]
- Inference of ancestral areas.[35]
- Molecular dating analysis.[36][37]
- Model dynamics of species diversification and extinction.[38]
- Elucidate patterns in pathogens dispersal.[39]
References
- ↑ Laplace, P. 1774. "Memoire sur la Probabilite des Causes par les Evenements." l'Academie Royale des Sciences, 6, 621-656. English translation by S.M. Stigler in 1986 as "Memoir on the Probability of the Causes of Events" in Statistical Science, 1(3), 359-378.
- ↑ Hastings,W.K. 1970. Monte Carlo sampling methods using Markov chains and their applications. Biometrika, 57, 97–109
- ↑ Metropolis, N., A. W. Rosenbluth, M. N. Rosenbluth, A. H. Teller, and E. Teller. 1953. Equation of state calculations by fast computing machines. J. Chem. Phys. 21:1087–1092.
- ↑ Felsenstein, J. 2004. Inferring phylogenies. Sinauer Associates, Sunderland, Massachusetts
- ↑ Yang, Z., Rannala, B. 1997. Bayesian phylogenetic inference using DNA sequences: a Markov Chain Monte Carlo Method. Mol. Biol. Evol. 14 (7) 717–24 http://mbe.oxfordjournals.org/cgi/pmidlookup?view=long&pmid=9214744 doi=10.1093/oxfordjournals.molbev.a025811
- ↑ Geyer,C.J. 1991. Markov chain Monte Carlo maximum likelihood. In Keramidas (ed.), Computing Science and Statistics: Proceedings of the 23rd Symposium on the Interface. Interface Foundation, Fairfax Station, pp. 156–163.
- ↑ Larget, B., and D. L. Simon. 1999. Markov chain Monte Carlo algorithms for the Bayesian analysis of phylogenetic trees. Mol. Biol. Evol. 16:750–759.
- ↑ Mau,B., Newton,M. and Larget,B. 1999. Bayesian phylogenetic inference via Markov chain Monte carlo methods. Biometrics, 55, 1–12.
- ↑ Felsenstein, J. 1978. Cases in which parsimony and compatibility methods will be positively misleading. Systematic Zoology 27 (4): 401–410. doi:10.1093/sysbio/27.4.401.
- ↑ Swofford, D., Olsen, G., Wadell, P. And Hillis, D.M. 1996. Phylogenetic inference. In Hillis, Moritz and Mable (eds), Molecular Systematics, 2nd edition. Sinauer, Surderland, MA, pp. 407-511.
- ↑ Garcia-Sandoval, R. 2014. Why some clades have low bootstrap frequencies and high Bayesian posterior probabilities. Israel Journal of Ecology & Evolution 60 (1): 41-44.
- ↑ Suzuki, Y. et al. 2002. Over credibility of molecular phylogenies obtained by Bayesian phylogenetics. Proc. Natl. Acad. Sci. U. S. A. 99, 16138–16143
- ↑ Erixon, P. et al. 2003. Reliability of Bayesian posterior probabilities and bootstrap frequencies in phylogenetics. Syst. Biol. 52, 665–673
- ↑ Nylander, J. A. A. 2004. MrModeltest 2.0. Program distributed by the author. Evolutionary Biology Centre, Uppsala University. Norbyvagen 18 D. SE-752 36, Uppsala, Sweden.
- ↑ Huelsenbeck, J. P. and F. Ronquist. 2001. MrBayes: Bayesian inference of phylogeny. Bioinformatics 17:754–755.
- ↑ Maddison,D.R., Swofford,D.L. And Maddison,W.P. 1997. NEXUS: an extensible file format for systematic information. Syst. Biol., 46: 590-621.
- ↑ Metropolis, N., A. W. Rosenbluth, M. N. Rosenbluth, A. H. Teller, and E. Teller. 1953. Equation of state calculations by fast computing machines. J. Chem. Phys. 21:1087–1092.
- ↑ Jukes, T.H. and Cantor, C.R. 1969. Evolution of Protein Molecules. New York: Academic Press. pp 21–132.
- ↑ Yang, Z. 1993. Maximum likelihood estimation of phylogeny from DNA sequences when substitutions rates differ over sites. Mol. Biol. Evol. 10: 1396-1401.
- ↑ Ronquist F., Huelsenbeck J.P. 2003. Mrbayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 19:1572–1
- ↑ Ronquist F., TeslenkoM.,Van Der Mark P.,Ayres D.L., DarlingA.,Hhna S., Larget B., Liu L., Suchard M.A., Huelsenbeck J. 2012. Mrbayes 3.2: Efficient bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61:539–542.
- ↑ Lord, E., Leclercq, M., Boc, A., Diallo, A.B., Makarenkov, V. 2012. Armadillo 1.1: An Original Workflow Platform for Designing and Conducting Phylogenetic Analysis and Simulations. PLoS ONE 7(1): e29903. doi:10.1371/journal.pone.0029903
- ↑ Suchard, M.A. and Redelings, B.D. 2006. BAli-Phy: simultaneous Bayesian inference of alignment and phylogeny, Bioinformatics. 22:2047-2048
- ↑ Wilson, I., Weale, D. and Balding, M. 2003. Inferences from DNA data: population histories, evolutionary processes and forensic match probabilities. Journal of the Royal Statistical Society: Series A (Statistics in Society), 166: 155-188
- ↑ Pagel, M. and Meade, A. 2006. Bayesian analysis of correlated evolution of discrete characters by reversible-jump Markov chain Monte Carlo. American Naturalist, 167, 808-825
- ↑ Lartillot N., Philippe H. 2004. A Bayesian Mixture Model for Across-Site Heterogeneities in the Amino-Acid Replacement Process. Molecular Biology and Evolution 21(6):1095-1109
- ↑ Drummond, A.J., Rambaut, A. 2007. Beast: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol. 7:214
- ↑ Ané, C., Larget, B., Baum, D.A.,Smith, S.D., Rokas, A. 2007. Bayesian estimation of concordance among gene trees. Molecular Biology and Evolution 24(2), 412-426
- ↑ Milne, I., Lindner, D., Bayer, M., Husmeier, D., McGuire, G., Marshall, D.F. and Wright, F. 2008. TOPALi v2: a rich graphical interface for evolutionary analyses of multiple alignments on HPC clusters and multi-core desktops. Bioinformatics 25 (1), 126-127
- ↑ Alonso, R., Crawford, A.J. & Bermingham, E. 2011. Molecular phylogeny of an endemic radiation of Cuban toads (Bufonidae: Peltophryne) based on mitochondrial and nuclear genes. Journal of Biogeography 39 (3): 434-451
- ↑ Antonelli, A., Sanmart.n, I. 2011. Mass Extinction, Gradual Cooling, or Rapid Radiation? Reconstructing the Spatiotemporal Evolution of the Ancient Angiosperm Genus Hedyosmum (Chloranthaceae) Using Empirical and Simulated Approaches. Syst. Biol. 60(5):596–615
- ↑ de Villemereuil, P.,Wells, J.A., Edwards, R.D. and Blomberg, S.P. 2012. Bayesian Phylogeography Finds Its Roots BMC Evolutionary Biology 12:102
- ↑ Ronquist, F. 2004. Bayesian inference of character Evolution. Trends in Ecology and Evolution 19 No.9: 475-481
- ↑ Schäffer , S., Koblmüller, S., Pfingstl, T., Sturmbauer, C., Krisper, G. 2010. Ancestral state reconstruction reveals multiple independent evolution of diagnostic morphological characters in the “Higher Oribatida” (Acari), conflicting with current classification schemes. BMC Evolutionary Biology 10:246
- ↑ Filipowicz, N., Renner, S. 2012. Brunfelsia (Solanaceae): A genus evenly divided between South America and radiations on Cuba and other Antillean islands. Molecular Phylogenetics and Evolution 64: 1-11
- ↑ Bacon, C.D., Baker, W.J., Simmons, M.P. 2012a. Miocene dispersal drives island radiations in the palm tribe Trachycarpeae (Arecaceae). Systematic Biology 61: 426–442
- ↑ Särkinen, T., Bohs, L., Olmstead,R.G. and Knapp, S. 2013. A phylogenetic framework for evolutionary study of the nightshades (Solanaceae): a dated 1000-tip tree. BMC Evolutionary Biology. 13:214
- ↑ Silvestro, D., Schnitzler, J., Liow, L.H., Antonelli, A., Salamin, N. 2014. Bayesian Estimation of Speciation and Extinction from Incomplete Fossil Occurrence Data. Syst. Biol. 63(3):349–367
- ↑ Lemey, P., Rambaut, A., Drummond, A.J., Suchard, M.A. 2009. Bayesian Phylogeography Finds Its Roots. PLoS Comput Biol 5(9): e1000520
External links
Topics in phylogenetics | |
|---|---|
| Relevant fields | |
| Basic concepts | |
| Inference methods | |
| Current topics | |
| Group traits | |
| Group types | |