2,4 Dienoyl-CoA reductase
| 2,4-dienoyl CoA reductase 1, mitochondrial | |
|---|---|
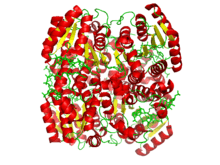 Crystallographic structure of a tetramer of the ternary complex of DECR1, hexanoyl-coenzyme A, and NADP.[1] | |
| Identifiers | |
| Symbol | DECR1 |
| Alt. symbols | DECR |
| Entrez | 1666 |
| HUGO | 2753 |
| OMIM | 222745 |
| PDB | 1w6u |
| RefSeq | NM_001359 |
| UniProt | Q16698 |
| Other data | |
| EC number | 1.3.1.34 |
| Locus | Chr. 8 q21.3 |
2,4 Dienoyl-CoA reductase also known as DECR1 is a protein which in humans is encoded by the DECR1 gene which resides on chromosome 8.[2][3][4]
This gene encodes an accessory enzyme that participates in the beta oxidation and metabolism of polyunsaturated fatty enoyl-CoA esters. Specifically, it catalyzes the reduction of 2,4 Dienoyl-CoA thioesters of varying length by NADPH cofactor to 3-trans-enoyl-CoA of equivalent length. Unlike the breakdown of saturated fat, cis and trans polyunsaturated fatty acid degradation requires three additional enzymes to generate a product compatible with the standard beta oxidation pathway. DECR is the second such enzyme (The others being Enoyl CoA isomerase and Dienoyl CoA isomerase) and is the rate limiting step in this auxiliary flow. DECR is capable of reducing both 2-trans,4-cis-dienoyl-CoA and 2-trans,4-trans-dienoyl-CoA thioesters,[5] as well as double bonds at odd carbon positions,[6] with equal efficiency. At this time, there is no clear explanation for this of lack of stereo-specificity.
Structure
Gene
The human DECR1 gene has 11 exons and resides on chromosome 8 at q21.3.[2] Sequence alignment indicates that there are five highly conserved acidic residues, one of which might act as a proton donor.[7]
Protein
Eukaryotic DECR exists in both the mitochondria (mDECR) and the peroxisome (pDECR, coded by gene DECR2). The enzymes from each organelle are homologous and part of the short-chain dehydrogenase/reductase SDR super-family. mDECR is 124kD consisting of 335 amino acids before posttranslational modification.[3] The secondary structure shares many of the motifs of SDR, including a Rossman fold for strong NADPH binding. The protein exists as a homotetramer in physiological environment, but has been shown to also form monomers and dimers in solution.[7]
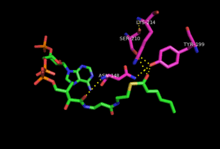
Crystallization of mDECR[1] shows the enzyme provides a network of hydrogen bonds from key residues in the active site to NADPH and the 2,4-dienoyl-CoA which positions the hydride at 3.4 Å to the Cδ, compared with 4.0 Å to the Cβ (not shown). The enolate intermediate discussed earlier is stabilized by residues additional hydrogen bonds to Tyr199 and Asn148. Lys214 and Ser210 (conserved residues in all SDR enzymes) are thought to increase the pKa of Tyr199 and stabilize the transition state.[1] Additionally, at one end of the active site there is a flexible loop that provides sufficient room for long carbon chains. This likely gives the enzyme flexibility to process fatty acid chains of various lengths. Substrate length for mDECR catalysis is thought to be limited at 20 carbons, at which the [very long chain fatty acid] is first partially oxidized by pDECR in the peroxisome.[8]
Enzyme Mechanism
Eukaryotic DECR
2,4 Dienoyl-CoA thioester reduction by NADPH to 3-Enoyl CoA occurs by a two-step sequential mechanism via an enolate intermediate.[9] DECR binds NADPH and the fatty acid thioester and positions them for specific hydride transfer to the Cδ on the hydrocarbon chain. The electrons from the Cγ-Cδ double bond move over to the Cβ-Cγ position, and those from the Cα-Cβ form an enolate. In the final step, a proton is abstracted from the water[10] to the Cα and the thioester is reformed, resulting in a single Cβ-Cγ trans double bond. Since the final proton comes from water, the pH has a significant effect on the catalytic rate with the enzyme demonstrating maximal activity at ~6.0.[7] A decrease in activity at pH < 6.0 can be explained by de-protonation of titratable residues that affect protein folding or substrate binding. Mutant proteins with modifications at key acidic amino acids (E154, E227, E276, D300, D117) show order of magnitude increases in Km and/or decreases in Vmax.

Prokaryotic DECR
2,4 Dienoyl-CoA Reductase from Escherichia Coli (E. Coli) shares very similar kinetic properties to that of eukaryotes, but differs significantly in both structure and mechanism. In addition to NADPH, E. Coli DECR requires a set of FAD, FMN and iron-sulfur cluster molecules to complete the electron transfer.[11] A further distinction is E. Coli DECR produces the final 2-trans-enoyl-CoA without the need for Enoyl CoA Isomerase.[10] The active site contains accurately positioned Tyr199 that donates a proton to the Cγ after hydride attack at the Cδ, completing the reduction in a single concerted step.[12] Surprisingly, mutation of the Tyr199 does not eliminate enzyme activity but instead changes the product to 3-trans-enoyl-CoA. The current explanation is that Glu164, an acidic residue in the active site, acts as a proton donor to Cα when Tyr199 is not present.[13]
Function
DECR is one of three auxiliary enzymes involved in a rate-limiting step of unsaturated fatty acid oxidation in mitochondria. In particular, this enzyme contributes to breaking the double bonds at all even-numbered positions, and some double bonds at odd-numbered position.[7] The structure of the ternary complex of pDCR (peroxisomal 2,4-dienoyl CoA reductases) with NADP and its substrate provides essential and unique insights into the mechanism of catalysis.[14] Unlike other members belonging to the SDR family, catalysis by pDCR does not involve a tyrosine-serine pair.[7] Instead, a catalytically critical aspartate, together with an invariant lysine, polarizes a water molecule to donate a proton for the formation of the product.[8] Although pDCR can use 2,4-hexadienoyl CoA as a substrate, the affinities for short chain fatty acids are lower. Analysis of the hinge movement of DCRs from the mitochondrion and peroxisomes sheds light on the reason behind the unique ability of the peroxisome to shorten very long chain fatty acids.[15]
Clinical Significance
Mutations in the DECR1 gene may result in 2,4 Dienoyl-CoA reductase deficiency,[16] a rare but lethal disorder. More recently, this deficiency has become a common disorder known to result in vomiting, low blood sugar, and even coma.
Due to its role in fatty acid oxidation, DECR may serve as a therapeutic target for treating non-insulin dependent diabetes mellitus (NIDDM), which features hyperglycemia due to increased fatty acid oxidation.[7]
In Knock-out mice studies,[17] DECR1−/− subjects accumulate significant concentrations of mono and polyunsaturated fatty acids in the liver during fasting (such as oleic acid, palmitoleic acid, linoleic acid, and linolenic acid). Mutant subjects were also found to have poor tolerance to cold, decrease in diurnal activity, and an overall reduction in adaptation to metabolic stressors.
See also
References
- 1 2 3 4 PDB: 1w6u; Alphey MS, Yu W, Byres E, Li D, Hunter WN (January 2005). "Structure and reactivity of human mitochondrial 2,4-dienoyl-CoA reductase: enzyme-ligand interactions in a distinctive short-chain reductase active site". J. Biol. Chem. 280 (4): 3068–77. doi:10.1074/jbc.M411069200. PMID 15531764.
- 1 2 "Entrez Gene: 2,4-dienoyl CoA reductase 1, mitochondrial".
- 1 2 Koivuranta KT, Hakkola EH, Hiltunen JK (December 1994). "Isolation and characterization of cDNA for human 120 kDa mitochondrial 2,4-dienoyl-coenzyme A reductase". Biochem. J. 304 (3): 787–92. PMC 1137403
 . PMID 7818482.
. PMID 7818482. - ↑ Helander HM, Koivuranta KT, Horelli-Kuitunen N, Palvimo JJ, Palotie A, Hiltunen JK (November 1997). "Molecular cloning and characterization of the human mitochondrial 2,4-dienoyl-CoA reductase gene (DECR)". Genomics. 46 (1): 112–9. doi:10.1006/geno.1997.5004. PMID 9403065.
- ↑ Cuebas D, Schulz H (December 1982). "Evidence for a modified pathway of linoleate degradation. Metabolism of 2,4-decadienoyl coenzyme A". J. Biol. Chem. 257 (23): 14140–4. PMID 7142199.
- ↑ Filppula SA, Yagi AI, Kilpeläinen SH, et al. (January 1998). "Delta3,5-delta2,4-dienoyl-CoA isomerase from rat liver. Molecular characterization". J. Biol. Chem. 273 (1): 349–55. doi:10.1074/jbc.273.1.349. PMID 9417087.
- 1 2 3 4 5 6 Yu W, Chu X, Chen G, Li D (February 2005). "Studies of human mitochondrial 2,4-dienoyl-CoA reductase". Arch. Biochem. Biophys. 434 (1): 195–200. doi:10.1016/j.abb.2004.10.018. PMID 15629123.
- 1 2 Hua T, Wu D, Ding W, Wang J, Shaw N, Liu ZJ (August 2012). "Studies of human 2,4-dienoyl CoA reductase shed new light on peroxisomal β-oxidation of unsaturated fatty acids". J. Biol. Chem. 287 (34): 28956–65. doi:10.1074/jbc.M112.385351. PMID 22745130.
- ↑ Fillgrove KL, Anderson VE (October 2001). "The mechanism of dienoyl-CoA reduction by 2,4-dienoyl-CoA reductase is stepwise: observation of a dienolate intermediate". Biochemistry. 40 (41): 12412–21. doi:10.1021/bi0111606. PMID 11591162.
- 1 2 Mizugaki M, Kimura C, Nishimaki T, Kawaguchi A, Okuda S, Yamanaka H (August 1983). "Studies on the metabolism of unsaturated fatty acids. XII. Reaction catalyzed by 2,4-dienoyl-CoA reductase of Escherichia coli". J. Biochem. 94 (2): 409–13. PMID 6355075.
- ↑ Liang X, Thorpe C, Schulz H (August 2000). "2,4-Dienoyl-CoA reductase from Escherichia coli is a novel iron-sulfur flavoprotein that functions in fatty acid beta-oxidation". Arch. Biochem. Biophys. 380 (2): 373–9. doi:10.1006/abbi.2000.1941. PMID 10933894.
- ↑ Hubbard PA, Liang X, Schulz H, Kim JJ (September 2003). "The crystal structure and reaction mechanism of Escherichia coli 2,4-dienoyl-CoA reductase". J. Biol. Chem. 278 (39): 37553–60. doi:10.1074/jbc.M304642200. PMID 12840019.
- ↑ Tu X, Hubbard PA, Kim JJ, Schulz H (January 2008). "Two distinct proton donors at the active site of Escherichia coli 2,4-dienoyl-CoA reductase are responsible for the formation of different products". Biochemistry. 47 (4): 1167–75. doi:10.1021/bi701235t. PMID 18171025.
- ↑ Ylianttila, MS; Pursiainen, NV; Haapalainen, AM; Juffer, AH; Poirier, Y; Hiltunen, JK; Glumoff, T (19 May 2006). "Crystal structure of yeast peroxisomal multifunctional enzyme: structural basis for substrate specificity of (3R)-hydroxyacyl-CoA dehydrogenase units.". Journal of Molecular Biology. 358 (5): 1286–95. doi:10.1016/j.jmb.2006.03.001. PMID 16574148.
- ↑ Emekli, U; Schneidman-Duhovny, D; Wolfson, HJ; Nussinov, R; Haliloglu, T (March 2008). "HingeProt: automated prediction of hinges in protein structures.". Proteins. 70 (4): 1219–27. doi:10.1002/prot.21613. PMID 17847101.
- ↑ Roe CR, Millington DS, Norwood DL, Kodo N, Sprecher H, Mohammed BS, Nada M, Schulz H, McVie R (May 1990). "2,4-Dienoyl-coenzyme A reductase deficiency: a possible new disorder of fatty acid oxidation". J. Clin. Invest. 85 (5): 1703–7. doi:10.1172/JCI114624. PMC 296625. PMID 2332510.
- ↑ Miinalainen IJ, Schmitz W, Huotari A, et al. (July 2009). "Mitochondrial 2,4-dienoyl-CoA reductase deficiency in mice results in severe hypoglycemia with stress intolerance and unimpaired ketogenesis". PLoS Genet. 5 (7): e1000543. doi:10.1371/journal.pgen.1000543. PMC 2697383. PMID 19578400.
External links
- 2,4-dienoyl-CoA reductase at the US National Library of Medicine Medical Subject Headings (MeSH)